【北京】导丝产品注册技术审评规范发布
来源: 北京市药品监督管理局
2022年11月30日 17:00
为指导和规范导丝产品的注册技术审评工作,帮助审评人员理解和掌握该类产品原理/机理、结构、性能、预期用途等内容,把握注册技术审评工作基本要求和尺度,对产品安全性、有效性作出系统评价,根据《医疗器械监督管理条例》(国务院令第739号)、《医疗器械注册与备案管理办法》(国家市场监督管理总局令第47号)、《医疗器械说明书和标签管理规定》(国家食品药品监督管理总局令第6号)、《医疗器械通用名称命名规则》(国家食品药品监督管理总局令第19号)、《国家药品监督管理局关于发布医疗器械产品技术要求编写指导原则的通告》(2022年第8号)、《国家药品监督管理总局关于发布医疗器械注册单元划分指导原则的通告》(2017年第187号)等相关规定,结合我市实际,北京市药品监督管理局组织制定了《导丝产品注册技术审评规范》,现予印发,自发布之日起施行。
本文著作权属原创者所有,不代表本站立场。我们转载此文出于传播更多资讯之目的,如涉著作权事宜请联系删除。
 加入会员
加入会员



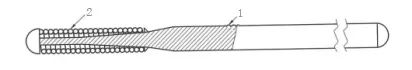 芯丝 2-绕丝
芯丝 2-绕丝

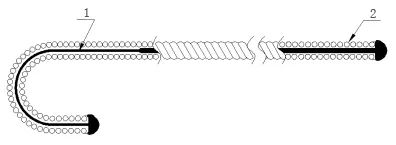

 导丝结构示例图5
导丝结构示例图5
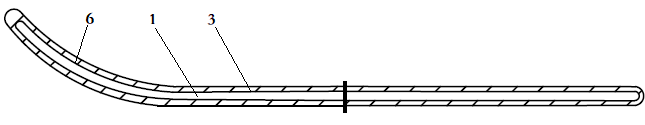 1-芯丝 3-聚合物护套 6-头端弯曲
1-芯丝 3-聚合物护套 6-头端弯曲


















