医械创新资讯


来源:本文刊登于《中国医疗器械信息》杂志2022年第28卷第19期
内容提要:美国FDA医疗器械审查标准转化是国内医疗器械注册技术审查指导原则的重要编制方式。文章以《类风湿因子检测试剂注册技术审查指导原则》为例介绍美国FDA医疗器械审查标准转化工作和转化成果情况,建议我国开展美国FDA医疗器械审查标准转化基于国内产品和监管实际情况,建立转化工作的指导和规范性文件,完善转化成果的修订和完善机制。
医疗器械技术审查指导原则的编制工作由国家药品监督管理局医疗器械技术审评中心组织开展,目前已建立了包括约500 项指导原则的文本库,以供开放下载和使用[1]。文本库内容包含有源医疗器械、无源医疗器械、体外诊断试剂类医疗器械技术审查指导原则和通用指导原则。其中有相当比例的指导原则编制工作是以美国食品药品管理局(Food and Drug Administration,FDA)医疗器械审查标准转化的方式进行的,如《家用体外诊断医疗器械注册技术审查指导原则》、《抗核抗体检测试剂注册技术审查指导原则》、《类风湿因子检测试剂注册技术审查指导原则》等。文章以《类风湿因子检测试剂注册技术审查指导原则》的编制工作为例,介绍我国开展美国FDA 医疗器械审查标准转化工作的基本情况,并对转化成果的应用及后续转化工作的开展提出建议。
1.概述
我国医疗器械产品注册相关法规体系构架的构成由上至下依次为行政法规、部门规章、规范性文件、技术指导原则和技术标准等。按照《体外诊断试剂注册与备案管理办法》[2](国家市场监督管理总局令第48 号)的要求,体外诊断试剂注册、备案应当遵守相关法律、法规、规章、强制性标准,遵循体外诊断试剂安全和性能基本原则,参照相关技术指导原则,证明注册、备案的体外诊断试剂安全、有效、质量可控,保证信息真实、准确、完整和可追溯,这为指导原则的应用确定了法规依据。
对各国医疗器械监管机构的政策法规对比研究是我国医疗器械监管科学研究的重要工作内容之一。截至2015 年,美国FDA发布的医疗器械审查标准已有700 多项,包括产品类和通用类的审查标准,覆盖面极其广泛[3,4]。选择其中重要的和我国医疗器械技术指导原则体系构建急需的审查标准进行合理转化,可以迅速为我国医疗器械监管工作补充技术性参考文件,为医疗器械技术审评提供审查依据,同时指导相应产品注册申请人的产品研发及注册申报资料的准备及撰写工作。2019年度第二类医疗器械注册技术审查指导原则编写计划中,共有30 个指导原则编制项目立项,其中6 项为美国FDA审查标准转化项目,占比达到20%。在监管对象层面,医疗器械研发团队及生产企业充分研究医疗器械相关国际政策法规及技术规范是全球化发展对其提出的必然要求。
2.《类风湿因子检测试剂注册技术审查指导原则》转化实例分析
2.1 背景及目的
伴随着环境的恶化和人口老龄化的趋势越来越严重,老年性疾病的发病率也逐年升高,类风湿性疾病的患者也越来越多。类风湿因子最初在类风湿关节炎血清中发现,后发现其在其他自身免疫性疾病及感染、淋巴细胞增生性疾病中能检测到[5,6]。类风湿因子的检测结果为临床医生的诊断和治疗提供着重要数据支持,同时,研究发现类风湿因子也是很多其他临床检测项目的内源性干扰物[7-10]。据统计,国内外关于类风湿因子检测情况的文献报道屡见不鲜,但目前我国尚且没有类风湿因子检测相关的技术文件对商品化的类风湿因子检测试剂产品进行规范和指导,国内各审评机构对该类产品的注册技术审评工作缺少统一尺度,这不利于行业内该类产品的整体质量控制,也不利于医疗机构检查检验结果互认工作的推进。为解决上述问题,课题组立项编制类风湿因子检测试剂注册技术审查指导原则,采用美国FDA 体外诊断器械审查标准转化的工作方式。
2.2 方法和过程
《类风湿因子测定试剂注册技术审查指导原则》转化工作自2019 年2 月启动到2020 年12 月发布实施,历时两年,工作内容及阶段性成果见表1。转化的主要研究依据包括美国FDA 医疗器械审查标准《Review Criteria for Assessment of Rheumatoid Factor (RF) in vitro Diagnostic Devices Using Enzyme-Linked Immunoassay (EIA), Enzyme Linked Immunosorbent Assay (ELISA), Particle Agglutination Tests, and Laser and Rate Nephelometry》(中文名称《使用酶联免疫测定(EIA)、酶联免疫吸附测定(ELISA)、颗粒凝集试验以及激光和速率比浊法评估类风湿因子(RF)体外诊断器械的审查标准》,以下简称“美国FDA审查标准”)、我国医疗器械注册法规、规范性文件和国内已上市类风湿因子检测试剂产品情况等,主要研究方法有文献检索、生产企业现场调研、问卷调研、数据研究、专题研讨会、研讨会、公开征求意见等。
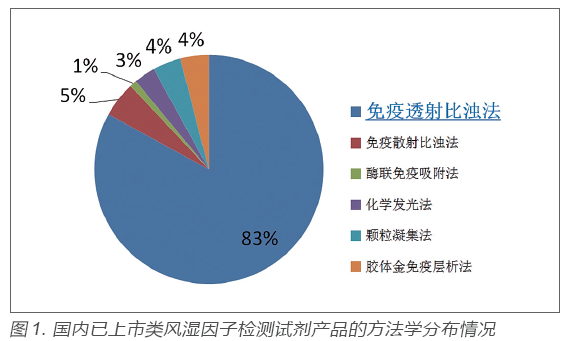
类风湿因子临床检验及临床应用情况调研结果显示,当前临床实验室检测类风湿因子最常用方法为(乳胶增强)免疫比浊法,包括免疫透射比浊法和免疫散射比浊法。
2.3.2 产品技术要求中性能指标及检验方法
目前国内尚且没有针对类风湿因子检测试剂产品专门的国家标准和行业标准。现场调研及问卷调研结果显示,已上市类风湿因子检测试剂产品的性能指标确定大多依据YY/T 1255-2015《免疫比浊法检测试剂(盒)(透射法)》。行业标准YY/T 1549-2017《生化分析仪用校准物》、YY/T 1662-2019《生化分析仪用质控物》、YY/T 1652-2019《体外诊断试剂用质控物通用技术要求》自发布实施以来,各省在产品注册过程中开始建议企业执行该推荐性行业标准,生产企业也逐步开始按照行业标准的要求进行性能研究和指标确定。因此,指导原则中提示了制造商可以参考执行标准YY/T 1255-2015、YY/T 1549-2017、YY/T 1662-2019、YY/T 1652-2019等相关标准的要求制定产品技术要求。
2.3.3 产品说明书中重点提示的内容
课题组根据调研及临床专题研讨会获得的类风湿因子检测试剂产品研发和临床使用过程中的关注内容,明确了说明书中类风湿因子检测试剂的适用范围、干扰、参考区间等重点关注内容。
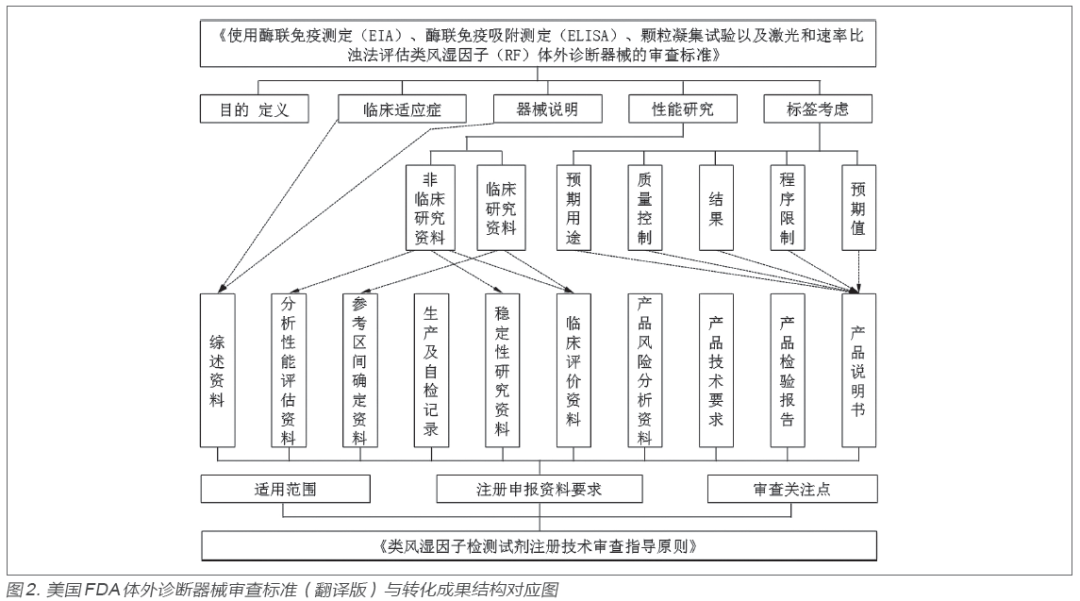
2.4 转化成果与美国FDA审查标准对比分析
经过历时两年的美国FDA审查标准转化,最终形成转化成果即《类风湿因子检测试剂注册技术审查指导原则》。该指导原则于2020 年12 月由国家药品监督管理局网站公开发布实施[11]。指导原则内容主要包括适用范围、注册申报资料要求、审查关注点等三部分,如图2 所示。