受理号:CQZ2101436
医疗器械产品注册技术审评报告
产品中文名称:吻合口加固修补片
国家药品监督管理局
医疗器械技术审评中心
基本信息
一、申请人名称
北京博辉瑞进生物科技有限公司
二、申请人住所
北京市大兴区中关村科技园区大兴生物医药产业基地药谷一号国际研发孵化园6#厂房西侧
北京市大兴区中关村科技园区大兴生物医药产业基地药谷一号国际研发孵化园6#厂房西侧;
北京市大兴区中关村科技园区大兴生物医药产业基地天富街9号10幢4层北侧
技术审评概述
吻合口加固修补片共包含管状型、平片型、圆型。管状型和圆型由修补片、背衬、牵引线三部分构成,平片型只有修补片。修补片由脱细胞猪小肠粘膜下层材料制备而成,与背衬用牵引线进行缝合固定。平片型无背衬和牵引线。产品经环氧乙烷灭菌,一次性使用。产品货架有效期24个月。
本产品配合吻合器用于吻合部位的加固。适用于远端胃切除术、近端胃切除术、袖状胃切除术、胃肠吻合术。
表1型号规格表
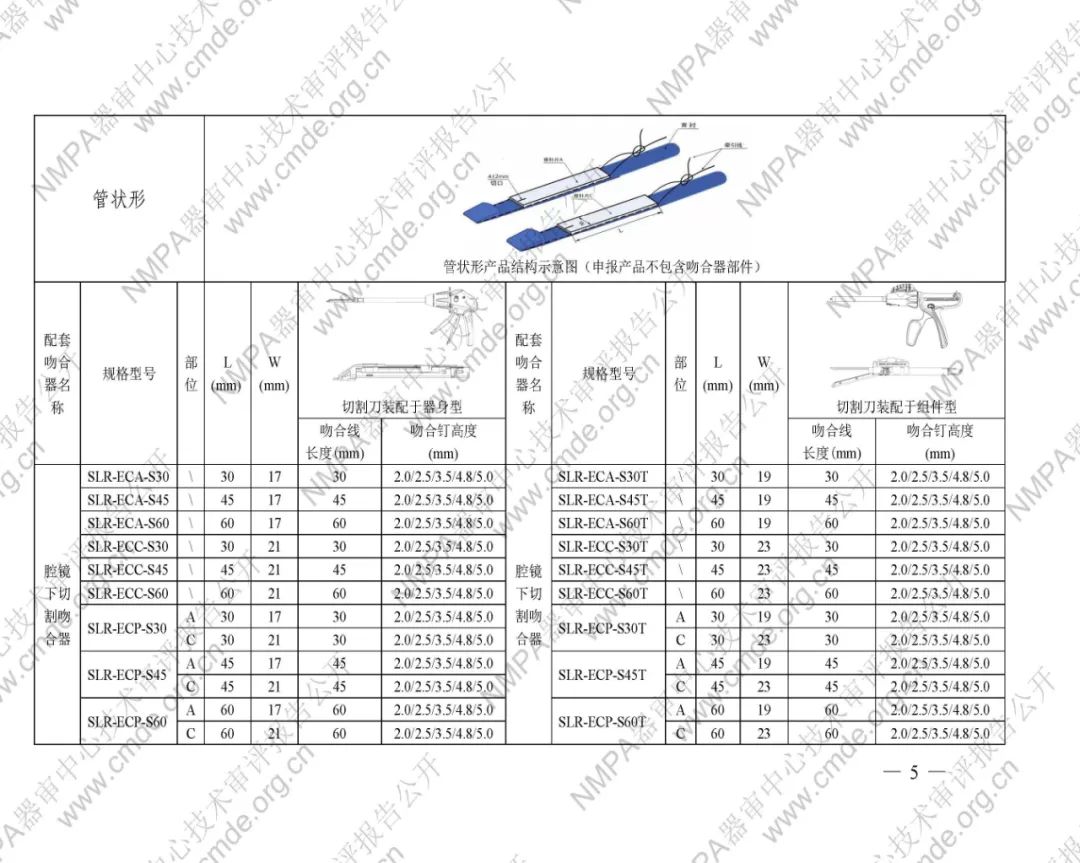
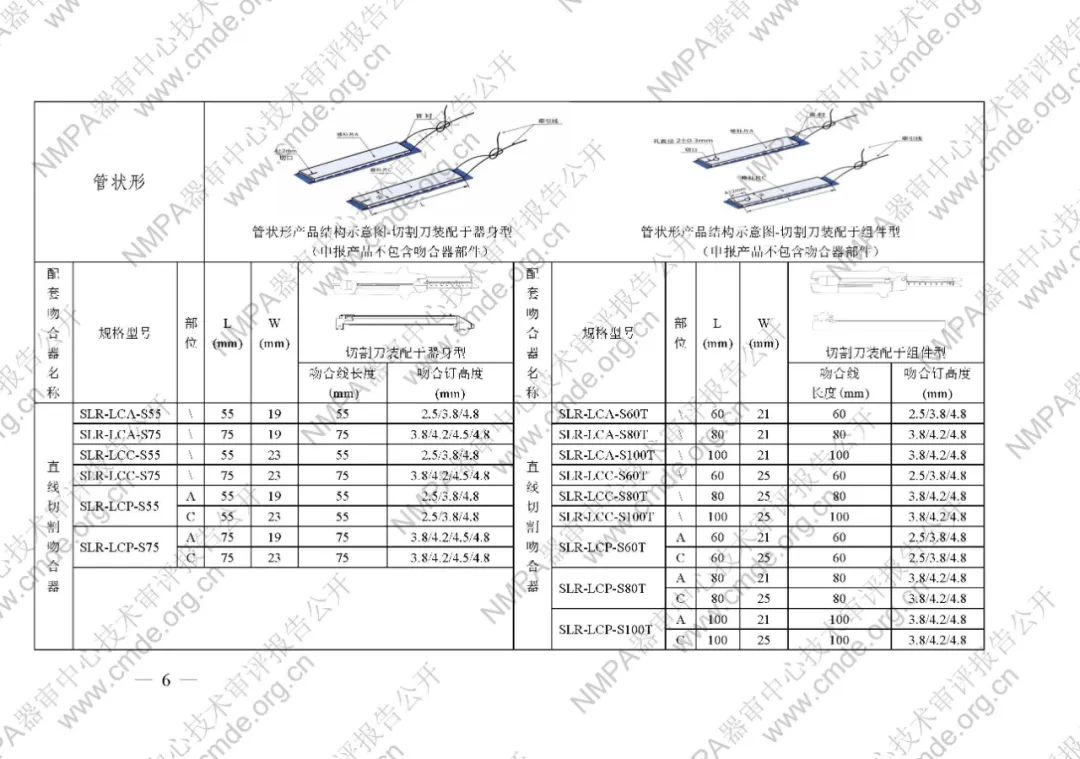
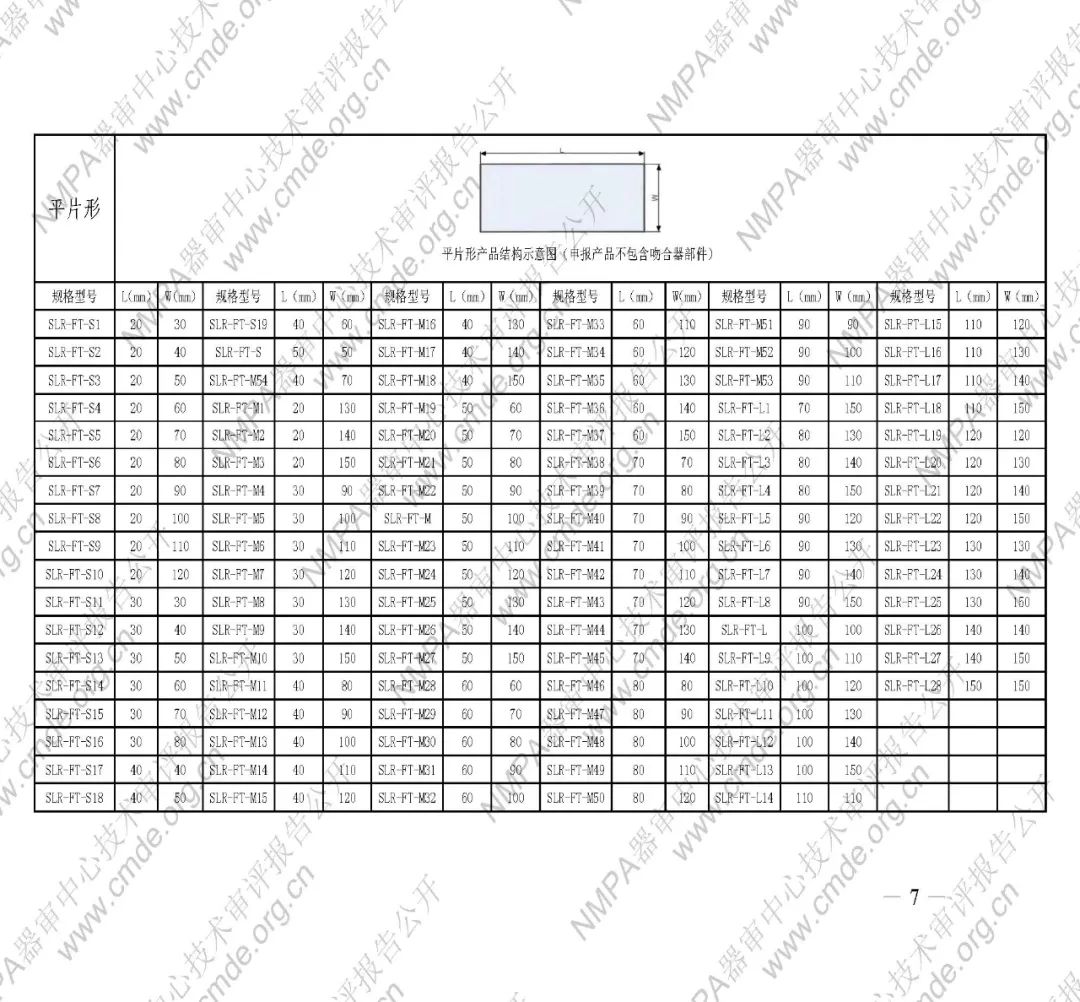
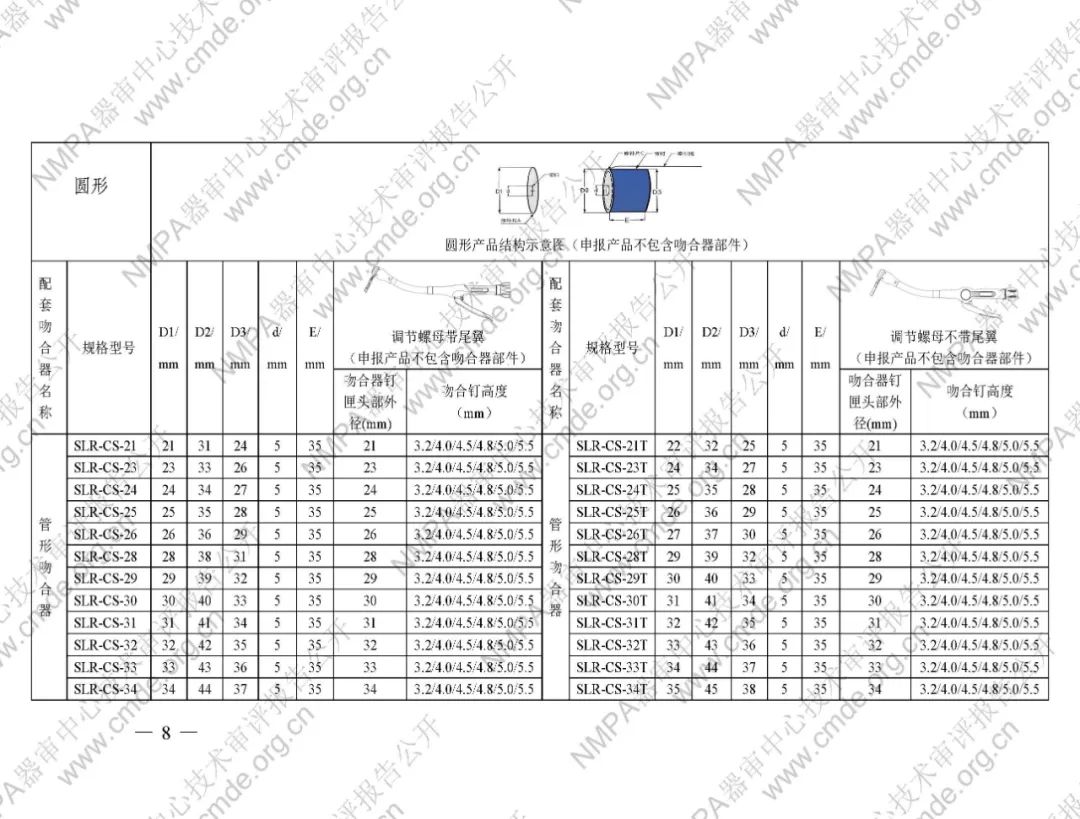
加固吻合部位,使组织断面均匀受压,分散钉孔处应力。
(一)产品性能研究1.产品技术要求研究产品技术要求研究项目如表2所示。
表2产品技术要求研究项目
产品性能评价包括:外观、尺寸、厚度、缝合强度、抗张强度、顶破强度、单位面积重量、拉伸伸长率、断裂伸长率、结构特性、可装配性、孔隙率、酸碱度、炽灼残渣、重金属、环氧乙烷残留量、过氧乙酸残留量、猪胰蛋白酶残留量、无菌、细菌内毒素、细胞残留检查、DNA残留量、修补片吸附性能的研究、修补片降解性能的研究。
该产品修补片为植入器械,与组织持久接触(大于30天)。申请人依据GB/T 16886系列标准进行了生物相容性评价,选择开展的生物学试验项目包括:热原、细胞毒性、皮内反应试验、迟发型超敏反应、急性全身毒性、亚慢性全身毒性、鼠伤寒沙门氏菌回复突变(Ames)试验、体外小鼠淋巴瘤细胞突变试验、染色体畸变试验、植入与降解,生物相容性风险可接受。部分型号含有的背衬和牵引线,与人体组织短期接触,申请人依据GB/T 16886系列标准进行了生物相容性评价,选择开展的生物学试验项目包括细胞毒性、皮内反应试验、迟发型超敏反应。综上,产品生物学风险可接受。
该产品为猪源性材料,申请人依据《动物源性医疗器械注册技术审查指导原则》和YY/T 0771动物源医疗器械系列标准提供生物安全性研究资料。
申请人按照YY/T 0771动物源医疗器械系列标准,对原材料来源、收集与处置的控制进行风险管理,对病毒灭活/去除工艺进行了验证。申请人还提供了免疫原性风险评价资料,参照GB/T 16886.20-2015《医疗器械生物学评价第20部分医疗器械免疫毒理学试验原则和方法》对本企业相同材质的其他产品开展了免疫毒性试验,结果表明,与阴性对照组相比,被测样品低、中、高剂量处理28天皆未对BALB/C小鼠免疫系统的功能造成不良影响;对α-Gal抗原清除率和DNA残留量进行检测,作为免疫原性质量控制的项目。
生物安全性研究资料显示产品的原料控制、病毒灭活、免疫原性和免疫毒性方面的风险可接受。
该产品采用环氧乙烷灭菌,无菌状态提供。申请人提供了灭菌确认报告,证明无菌保证水平可达10-6。环氧乙烷残留量不大于10μg/g,2-氯乙醇残留量不超过5mg/cm2。
该产品货架有效期为两年。申请人提供了货架有效期验证报告,验证方式为加速老化和实时老化验证,包括产品稳定性、包装完整性和运输模拟验证资料。
申请人开展了动物试验研究,通过试验用巴马小型猪为试验系统建立胃肠吻合动物模型,评价吻合口加固修补片在胃肠吻合口加固修补术中的安全性和有效性,观察的指标包括临床观察、血液学观察、术中疗效观察、大体解剖观察、组织病理学观察。研究结果表明,吻合口加固修补片90d时完全降解,使用吻合口加固修补片进行胃肠吻合,对吻合口有加固、止血的作用。供试品组与空白对照组动物的表现无明显差异,组织病理学无可见病变。
申请人采用临床试验的方式进行临床评价,临床试验的目的为评价申报产品用于胃部分切除吻合的软组织的安全有效性。临床试验为多中心、随机对照的优效性设计。临床试验在5家临床试验机构进行,共入组了137例受试者。试验组为吻合口加固片配合吻合器进行手术,对照组为仅使用吻合器进行手术。主要评价指标为吻合器击发完成切割吻合10min内需要临床止血处置的单位长度吻合口出血点数目(观察部位:十二指肠残端、胃切除断端、胃空肠吻合部位);次要有效性评价指标为吻合器击发完成切割吻合至止血处理完毕后3min内吻合口的出血量及出血点处理时间、手术时间、吻合器操作性能参数等;安全性评价指标包括术后并发症发生情况(吻合口出血、吻合口漏、吻合口狭窄、腹腔感染、免疫排斥反应)和不良事件发生情况。
实际入组137例受试者,其中FAS集与SS集为137例(实验组69例,对照组68例),PPS集为128例(实验组65例,对照组63例)。主要评价指标:吻合器击发完成切割吻合10min内需要临床止血处置的单位长度吻合口出血点数目FAS集实验组为0.09±0.10个,对照组为0.16±0.14个,两组差值95%置信区间为(-0.113,-0.030),其上限小于0;PPS集实验组为0.08±0.09个,对照组为0.17±0.14个,两组差值95%置信区间为(-0.130,-0.046),其上限小于0。次要评价指标吻合器击发完成切割吻合至止血处理完毕后3min内吻合口的出血量及出血点处理时间试验组与对照组有统计学差异(P<0.05)。不良事件发生率与严重不良事件发生率两组之间无统计学意义(P>0.05)。
综上所述,结论显示该产品的安全性和有效性可以达到制造商的预期要求,满足临床需求。
(一)该产品临床使用为适用人群带来的主要受益为:配合吻合器用于吻合部位的加固。适用于远端胃切除术、近端胃切除术、袖状胃切除术、胃肠吻合术。
(二)该产品临床使用可能为适用人群带来的主要风险为:
本产品使用猪小肠粘膜下层组织材料,经病毒灭活、免疫原去除、成型、干燥、灭菌等工序制备而成,虽然去除了绝大部分的免疫原物质(细胞成分、动物源DNA和α-GAL抗原等),但残留的微量免疫原物质可能会导致相应的风险。从材料安全性的角度考虑,该产品在生产过程中对所有的风险源采取了控制措施,使所有风险处于可接受准则的范围内。
产品在某些非预期情况下,如未经过专业培训的使用者操作、可能会因使用操作错误导致发生治疗错误的风险。
(三)根据申请人提供的申报资料,经综合评价,在目前认知水平上,认为该产品的上市为适用人群带来的受益大于风险。但为保证用械安全,基于对主要剩余风险的规避,需在说明书中提示以下信息:
背衬和牵引线务必撤出体外废弃,体内不要留下除修补片以外的东西,如果背衬材料被夹住时,请用剪刀等进行分离,务必将背衬和牵引线取出体外废弃。
不可使用在需要长期才能愈合的部位。另外,不能在体内作永久性的固定作用。
作加固修补时,不要在有过度张力和负荷的部位使用,也不要在施加负荷的情况下使用。(有可能会引起破损和损伤)。
综合评价意见
本申报产品属于创新医疗器械(创新编号:CQTS1700209)。依据《医疗器械监督管理条例》(国务院令第739号)、《医疗器械注册与备案管理办法》(国家市场监督管理总局令2021年第47号)等相关医疗器械法规与配套规章,经对申请人提交的注册申报资料进行系统评价,申报产品符合安全性、有效性的要求,符合现有认知水平,建议准予注册。
2022年07月27日





