受理号:JQZ2100332
医疗器械产品注册技术审评报告
产品英文(原文)名称:Shockwave Coronary Intravascular Lithotripsy (IVL) Catheter
申请人名称:Shockwave Medical, Inc.
基本信息
一、申请人名称
Shockwave Medical, Inc.
二、申请人住所
5403 Betsy Ross Drive Santa Clara, CA USA 95054
三、生产地址
5403 Betsy Ross Drive Santa Clara, CA USA 95054
技术审评概述
产品由球囊、导管轴、导管座、充盈端口、导丝出口、连接器和脉冲发射器组成。产品为电子束灭菌,一次性使用,货架有效期2年。
产品在医疗机构使用,与本公司生产的血管内冲击波治疗设备(型号:IVLGCCD)配合,用于成人患者在支架植入术前对原发性冠状动脉的钙化病变(冠状动脉狭窄程度≥50%)进行预处理及球囊扩张。
C2IVL2512、C2IVL3012、C2IVL3512、C2IVL4012
该产品为冠脉血管用的一次性使用血管内冲击波导管,预期与本公司生产的血管内冲击波治疗设备配合使用,用于对冠脉血管钙化病变进行处理和扩张。
产品基于体外碎石冲击波原理改进而来,将聚焦式高强度能量改为发散式低强度能量,通过血管内导管方式导入外周血管对钙化病变部位进行松解,以缓解血管狭窄程度或便于后续治疗。目前尚无同样原理和使用方式的产品在我国获准注册。
产品导管上含有若干液电式能量波源(脉冲发射器),通过接受发生器传递的电能转化为机械能向外输出冲击波能量。能量波源外部含有球囊,与球囊加压装置配合使用可用于扩张狭窄部位血管,球囊内部可注入生理盐水/造影剂的1:1混合液用于传递冲击波能量。
申请人提供了产品性能研究资料以及产品技术要求的研究和编制说明,给出了物理性能、机械性能、化学性能、球囊性能、声输出性能、电气安全和电磁兼容等功能性、安全性指标以及与质量控制相关的其他指标的确定依据。产品技术要求中各指标参考了相关的国家、行业标准,包括:GB 9706.1-2007、YY 0505-2012、YY 0285.1-2017、YY 0285.4-2017等。
申请人依据GB/T 16886.1-2011对成品中与患者直接接触的导管的生物相容性进行了评价。所评价材料短时接触人体血路,实施了生物学试验(细胞毒性、致敏、皮内反应、急性全身毒性、热原、遗传毒性、血液相容性),提交了境外检测机构出具的生物学试验报告。导管外表面含有涂层,属于常规材料,提供了材料使用安全史和毒理学分析的相关资料。
产品由生产企业委托第三方机构进行电子束灭菌,无菌保证水平为10-6,申请人依据相关标准对产品进行VDmax25灭菌确认,提交了剂量分布验证和灭菌负载验证资料,以及相应的灭菌效果确认报告。辐照灭菌方式不涉及残留毒性研究。
产品为一次性使用,货架有效期2年。申请人对产品进行加速老化试验,并对老化后产品进行性能测试和包装验证。基于相关标准进行了存储运输试验。
申请人提供了产品基于猪体内模型开展的慢性动物试验对照研究资料,评价产品将冲击波能量输送到冠脉血管的安全性和血管效应。对共计6只动物进行模拟冲击波治疗,选择导管最大耐受能量对各动物个体的主要冠脉血管部位进行试验,随后置入支架,治疗后随访28天。结果显示,全部动物均存活至随访期末,治疗过程中未出现并发症,各治疗部位影像学检查未发现明确管腔狭窄或血流不畅,病理学检查显示治疗部位组织和正常组织形态相似,动物体征健康。
申请人提供了产品的冲击波声压能量分布研究测试报告。模拟实际临床使用状态,使用1:1混合液在规定气压条件下充盈球囊,分别测试不同型号导管的输出特性,测量并计算每个单一波源在球囊界面方向上的声压传输范围,以及所有波源能量叠加后的覆盖区域。结果证实产品冲击波能量范围和强度可涵盖预期治疗部位。
申请人还提供了产品球囊爆破压和耐受性的性能验证资料;提供了基于石膏模型的冲击波作用于模拟钙化病变的碎裂有效期验证资料。
产品符合医用电气相关通用安全标准(GB 9706.1-2007)和并列安全标准(YY 0505-2012)的要求,提供由医疗器械检验机构出具的符合标准要求的检验报告。
申请人采用临床试验路径进行临床评价,提交了境外临床试验数据。临床试验目的为评价一次性使用冠脉血管内冲击波导管在支架植入前用于治疗冠状动脉钙化病变的安全性与有效性。提交的临床数据包括Disrupt CAD I、II、III,其中Disrupt CAD III为IDE研究,Disrupt CAD I为上市前临床试验,Disrupt CAD II为欧洲上市后临床研究。
表1临床研究概况
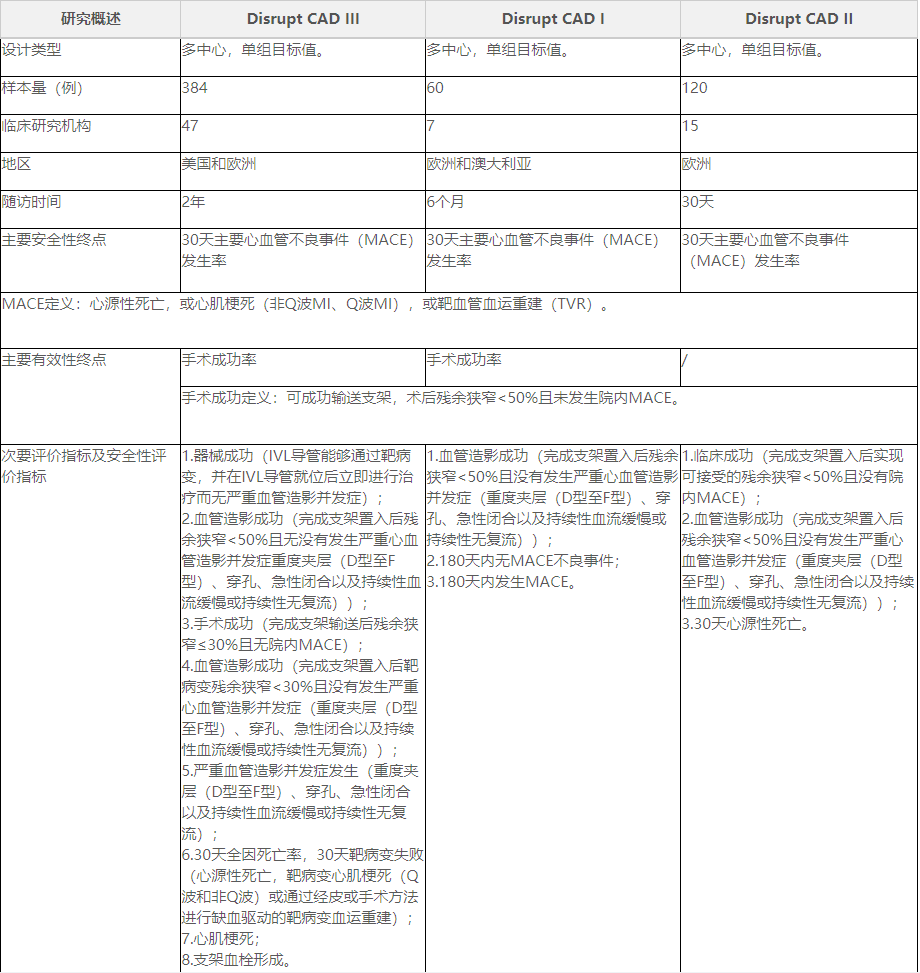
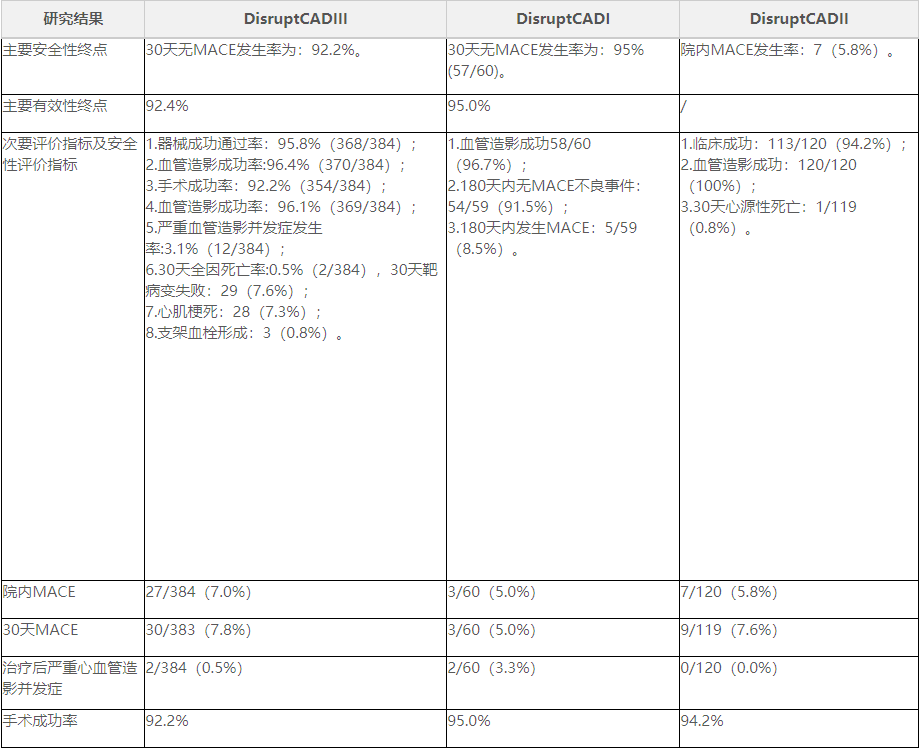
表2临床研究结果(二)临床研究结果(见表2)。
该产品用于成人患者在支架植入术前对原发性冠状动脉钙化病变(冠状动脉狭窄程度≥50%)进行预处理及球囊扩张,主要受益是为患者提供了一种额外的钙化病变处理手段。主要风险为临床使用时的手术相关风险,特别是冲击波可能导致的钙化斑块脱落风险,以及不必要的使用或者操作不当等。四、产品受益风险判定
根据申请人提供的申报资料,经综合评价,在目前认知水平上,认为该产品的上市为适用人群带来的受益大于风险。
综合评价意见
该产品为优先审批医疗器械,首个申报的同类医疗器械产品。依据《医疗器械监督管理条例》(国务院令第680号)、《医疗器械注册管理办法》(原国家食品药品监督管理总局令第4号)等相关医疗器械法规与配套规章,经对申请人提交的注册申报资料进行系统评价,申报产品符合安全性、有效性的要求,符合现有认知水平,建议准予注册。






