受理号:CSZ2200011
体外诊断试剂产品注册技术审评报告
产品中文名称:α 和 β 地中海贫血基因检测试剂盒 (联合探针锚定聚合测序法)
产品管理类别:第三类
申请人名 称:华大生物科技(武汉)有限公司
国家药品监督管理局
医疗器械技术审评中心
基本信息
二、申请人住所 武汉市东湖新技术开发区高新大道 666 号武汉国家生物 产业基地项目 B、C、D 区研发楼 B2 栋
三、生产地址 武汉市东湖新技术开发区高新大道 666 号武汉国家生物产 业基地项目 B、C、D 区研发楼 B2 栋五楼、B1 栋一楼
技术审评概述
一、产品概述
表 1 主要组成成分


(二)产品预期用途
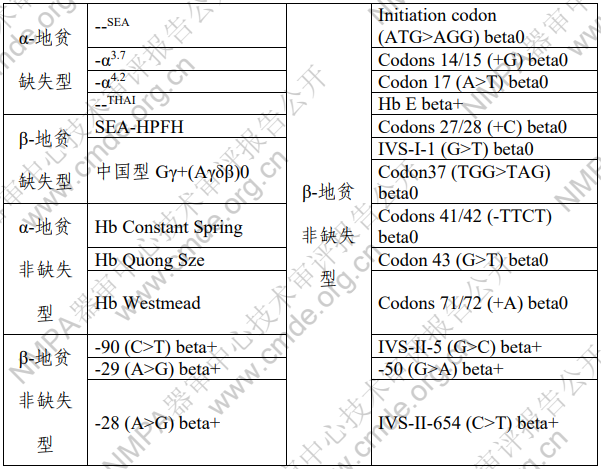
本试剂盒用于文库构建,采用高通量测序技术(联合探 针锚定聚合测序法)定性检测人外周血样本中基因组 DNA的 4 种 α-地贫缺失型、2 种 β-地贫缺失型、3 种 α-地贫非缺失 型和 16 种 β-地贫非缺失型。用于 α 和/或 β 地贫的辅助诊断(遗传诊断)。本试剂盒检测结果不作为地中海贫血临床诊 断的唯一依据,仅供临床参考。
表 2 试剂盒检测的地中海贫血基因变异信息
(三)产品包装规格
(四)产品检验原理
根据 α-地贫和 β-地贫发生相关的 HBA1、HBA2 和 HBB 三个基因的特点设计一系列引物,以实现基因的有效扩增。利用多重 PCR 技术将样本中的 HBA1、HBA2 和 HBB 基因 扩增富集并同时引入用于样本识别的标签序列。将多例(≤96 例)样本的 PCR 产物按一定比例混合成一个文库,经过 一系列的文库制备过程,每个文库中的样本 DNA 序列都加 上了用于测序及文库识别的接头序列。将制备好的文库等质 量比例上机测序,使用基因测序仪通过联合探针锚定聚合测 序技术对各文库中的样本 DNA 进行序列信息读取。每一条 DNA 的测序结果经过文库接头序列、样本标签序列和引物序 列比对拆分后将被精确定位到每一个样本的每一个扩增区 域中。
将每个样本的目标区域的测序结果与以 GRCh37 基因 组序列为参考构建的目标参考序列进行比对分析,再通过变 异检测、阳性判断值以及来自于 HbVar(2017 年 10 月版) 和 Ithanet 数据库(2017 年 10 月版)的变异坐标信息与变异 名称信息,判断样本中是否存在地贫基因变异以及何种变异。
二、临床前研究概述
试剂盒主要原材料包括:PCR 标签引物、PCR 反应液、 PCR 反应热启动酶、测序接头、末端修复缓冲液、T4 多聚 核苷酸磷酸激酶、T4 DNA 聚合酶 (rTaq 聚合酶)、Klenow Fragment、加“A”酶、连接酶、磁珠、阳性对照品、阴性对照 品等。主要原材料均为外购方式获得。PCR 标签引物和测序 接头为申请人自行设计后由专业的合成公司合成。
申请人选择有资质的供应商提供原料,通过功能性试验筛选出最佳原 材料和供应商,制定了各主要原材料质量标准并经检验合格。
企业参考品包括阳性参考品、阴性参考品、重复性参考 品、精密度参考品、检测限参考品。阳性参考品 37 份,由外 周血样本和细胞系样本组成,样本涵盖该产品可检出的所有 变异类型的杂合型;阴性参考品 14 份,由外周血样本和细胞 系样本组成,包含无变异样本、产品检测范围外的地贫变异 类型样本及同源序列变异样本;重复性参考品 5 份,均为细 胞系样本,涵盖产品可检出的 4 种代表性变异类型和 1 份无 变异的阴性样本;精密度参考品 5 份,为包含弱阳性浓度水 平在内不同浓度水平和无变异的外周血样本;检测限参考品 4 份,均为细胞系样本,每份检测限参考品做高、中、低 3 个 水平的梯度稀释,涵盖该产品可检出的 4 种代表性变异类型;检测限参考品 37 份,由细胞系样本和外周血样本组成,包含 产品可检出的所有变异类型的最低检出限水平。
试剂盒包含 4 份阳性对照品和 1 份阴性对照品,用于检 测过程的质量控制。阴阳性对照品均为细胞系样本,阳性对 照品涵盖该产品可检出的 4 种代表性变异类型,阴性对照品 为无变异样本。
申请人通过对多重 PCR 反应引物配比、PCR 反应标签 引物用量、PCR 反应体系和反应条件、PCR 产物混合比例、 阳性对照检测方式、文库制备起始量、纯化磁珠用量、打断 参数、末端修复反应体系、末端修复反应时间、加 A 反应体 系、连接反应体系、筛选磁珠用量、样本有效序列数、上机 文库数等条件进行筛选和优化,通过功能性试验,最终确定 了最佳的生产工艺与反应体系。
分析性能评估包括核酸提取试剂性能研究、准确度、重 复性、精密度、分析特异性、分析灵敏度等性能评估。针对核酸提取试剂性能研究,申请人采用临床样本平行 比较了核酸纯化试剂和核酸提取试剂的提取性能,通过对比 提取所得 DNA 的浓度和纯度,确定核酸提取试剂为该产品 的配套提取试剂。
准确度评估中,申请人分别在两种适配机型(基因测序 仪(MGISEQ-2000)和基因测序仪(MGISEQ-200))上使用 三批试剂盒对企业参考品盘中的 37 份阳性参考品进行检测, 阳性符合率为 100%。另外,申请人分别在两种适配机型(基 因测序仪(MGISEQ-2000)和基因测序仪(MGISEQ-200)) 上使用一批试剂盒对已知临床检测结果的临床样本进行检 测,试剂盒检测范围内各变异类型的阳性符合率、阴性符合率和总符合率均为 100%。精密度评估中,申请人使用三批试剂盒对临界浓度 10 ng/μL 和中高浓度 15 ng/μL 的 5 份外周血样本和 1 份细胞系 样本在两个实验室由两组操作者在不同适配机型(基因测序 仪(MGISEQ-2000)和基因测序仪(MGISEQ-200))的不同 仪器上进行检测,每个浓度的每个样本重复检测 3 次,连续 检测 20 天。检测结果均与已知结果一致。分析特异性评估中,申请人分别在两种适配机型(基因 测序仪(MGISEQ-2000)和基因测序仪(MGISEQ-200))上使用三批试剂盒对企业参考品盘中的 14 份阴性参考品进行 检测,阴性符合率为 100%;对 Hb Q-Thailand、Hb New York、 Hb J-Bangkok 等具有同源性序列的血红蛋白异常样本和-- FIL、-α21.9、Hb Lepore-Boston-Washington 等检测范围外的 地贫变异阳性样本进行检测,检测结果均为未检出检测范围 内的基因变异;对梯度稀释的血红素、甘油三酯、胆红素、 肝素钠、EDTA•K2、甲磺酸去铁胺、地拉罗司分散片、叶酸 片和爱乐维(复合维生素片)、布洛芬、盐酸氨溴索、蒲地蓝 消炎胶囊、头孢克肟干扰样本进行检测,检测结果表明,当 DNA 样本中血红素浓度≤200g/L、甘油三酯浓度≤1000 mg/dL、胆红素浓度≤2 mg/dL、EDTA•K2 浓度≤2.5 mg/mL、 甲磺酸去铁胺浓度≤1.6 mg/mL、地拉罗司浓度≤1.6 mg/mL、叶酸浓度≤0.38 mg/L、爱乐维(复合维生素)平均浓度 ≤35.6 mg/L、布洛芬浓度≤0.143mg/mL、盐酸氨溴索浓度≤ 0.12mg/mL、头孢克肟浓度≤0.286mg/mL、蒲地蓝消炎胶囊 浓度≤2.86mg/mL 时,干扰物质对试剂盒检测结果无明显影响,试剂盒具有较好的抗干扰能力。当样本中的肝素钠浓度 在 2IU/mL 以上时,试剂盒检测失败率高,试剂盒不适用于 检测肝素钠抗凝采血管采集的样本,样本采集应选用 EDTA•K2 抗凝采血管。最低检测限研究中,申请人分别在两种适配机型(基因 测序仪(MGISEQ-2000)和基因测序仪(MGISEQ-200))上 进行了研究。
首先选择产品可检出的 4 种代表性变异类型的 细胞系样本进行梯度稀释,每个浓度梯度样本重复检测 3 次, 将具有 100%检出率的最低浓度设定为预估最低检测限。之 后,将包含所有申报位点的杂合型样本稀释至预估最低检测 限浓度,每个样本重复检测 20 次,将检测结果为相应的基因 型别且每个型别的检出率均≥95%时浓度,设定为最低检测 限浓度。最后,申请人使用三批试剂盒对各个申报位点的杂 合型样本进行重复检测 20 次,验证了最低检测限。(四)阳性判断值
申请人使用该产品对 329 例已有地贫基因检测结果的临床样本进行检测,对非缺失型的阳性样本进行突变频率的统计分析,基于理论杂合突变频率和纯合突变频率,利用 Tukey’s 检验确定非缺失型的阳性判断值;对缺失型阳性样本 和阴性样本进行归一化序列的统计分析,利用 k-medians 聚 类算法确定缺失型的阳性判断值。
申请人使用该产品对 428 例已有地贫基因检测结果的临 床样本进行阳性判断值的验证。结果显示阳性符合率、阴性 符合率和总符合率均为 100%,Kappa 系数均为 1。建立的阳 性判断值能准确判断检测范围内的 25 种地贫变异类型的阴、 阳性。
为考核该产品在不同条件下性能的稳定性状况,为试剂 盒的包装、运输、使用和保存条件的确定和有效期的建立提 供依据,申请人进行了稳定性研究,包括效期稳定性、冻融 稳定性等。研究结果表明:包装盒 1 与包装盒 2 置于-18℃以 下保存,包装盒 3 置于 2℃~8℃保存,有效期为 12 个月;包装盒 1、包装盒 2 冻融 6 次仍可有效检测;试剂盒每天开 瓶 1 次连续开瓶 7 天、每 2 天开瓶 1 次连续 2 周、每周开瓶 1 次连续 4 周、每月开瓶 1 次 4 个月内开瓶小于 4 次仍可有 效检测。
适用样本稳定性研究:为考察该产品对不同条件下储存 不同时间的样本的检出性能,申请人使用该产品分别对短期储存、长期储存的样本进行测试,最终确定外周血样本在室 温下储存 9 天、在 2℃~8℃储存 14 天、在-18℃以下储存 9 个月仍可有效检测;样本反复冻融次数应不超过 6 次;样本 在 2℃~8℃运输时,运输时间不超过 8 天。
三、临床评价概述
申请人在郴州市第一人民医院、珠海市妇幼保健院、云 南省第一人民医院共三家临床机构完成了临床试验。针对已 有同类产品产品上市的基因型,采用试验体外诊断试剂与已 上市同类产品进行比较研究;针对尚无同类产品上市的基因 型,采用试验体外诊断试剂与 Sanger 测序法进行比较研究。
临床试验的样本类型为外周血。入组病例包括具有地贫症状 和/或体征的疑似地贫患者、血液学表现为小细胞低色素的疑 似地贫患者、临床考虑为地贫携带者、需与地贫进行鉴别诊 断的其他疾病患者等。对于尚无同类产品上市的基因型,提 交了支持该基因型临床意义的临床试验资料及相关文献。临床试验共入组受试者 1108 例。有效样本中阳性样本 1034 例。各基因型别的阳性样本情况如下:α-地贫缺失型 434 例,β-地贫缺失型 7 例,α-地贫非缺失型 252 例,β-地贫非缺 失型 723 例。
临床试验结果显示,该产品检测范围内的 25 个基因型 别的检测结果与对比试剂/Sanger 测序法相比的阳性符合率均为 100%、阴性符合率均为 100%、总符合率均为 100%。综上所述,该产品临床试验试剂符合《体外诊断试剂临 床试验技术指导原则》和《地中海贫血相关基因检测试剂注 册技术审查指导原则》的相关要求。临床试验结果显示该产 品与已上市同类产品一致性较好。
四、产品收益风险判定
根据申请人提供的申报资料,经综合评价,在目前认知 水平上,认为该产品的上市为适用人群带来的受益大于风险。但为保证用械安全,基于对主要剩余风险的规避,需要在说 明书中提示以下信息:1.本试剂盒的检测结果仅供临床参考,不能作为诊断的 唯一依据。对患者的临床诊断应结合临床症状/体征、病史、 其他实验室检查等情况综合考虑;2.本试剂盒适用于人的外周血样本检测,不适用于其它 样本检测;3.本试剂盒可以对 4 种缺失型 α-地中海贫血、2 种缺失 型 β-地中海贫血、3 种非缺失型 α-地中海贫血和 16 种非缺 失型 β-地中海贫血进行检测,但仍有检测范围之外的变异, 可能造成漏检,这类样本可用其他方法进一步验证。4.不合理的样本采集、运送、处理、核酸过度降解以及未经 验证的其他干扰或 PCR 抑制因子等均有可能会导致假阴性 或假阳性结果。
综合评价意见
本申报项目为境内第三类医疗器械产品注册,申请人的 注册申报资料符合现行要求,依据《医疗器械监督管理条例》 (国务院令第 739 号)、《体外诊断试剂注册与备案管理办法》 (国家市场监督管理总局令第 48 号)等相关医疗器械法规 与配套规章,经系统评价后,建议准予注册。






