


医械创新资讯

体外诊断试剂产品注册技术审评报告
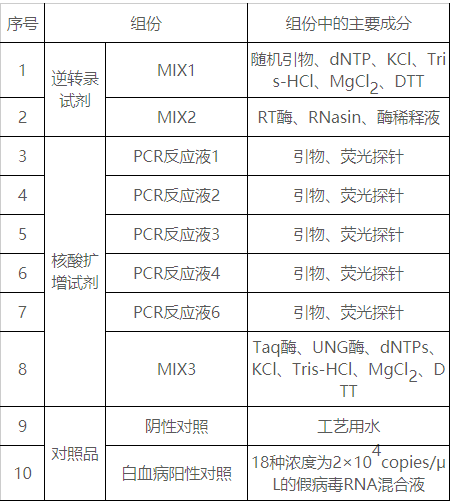
表1试剂盒主要组成成分
(二)产品预期用途
本试剂盒用于体外定性检测急性髓系白血病(AML)、急性淋巴细胞白血病(ALL)、以及其他混合表型白血病和前期诊断未明确分型的白血病患者骨髓样本中的BCR-ABL、SIL-TAL1、E2A-HLF、TEL-AML1、MLL-AF4、E2A-PBX1、AML1-ETO、MLL-AF9、PML-RARα、MLL-AF6、MLL-AF10、MLL-ELL、MLL-ENL、CBFβ-MYH11和DEK-CAN共15种白血病相关融合基因(其中MLL-AF6、MLL-AF10、MLL-ELL和MLL-ENL四种融合基因不能鉴别分型)。
本产品用于辅助白血病诊断与分型、临床药物的选择及评估疾病预后。
本产品通过对临床骨髓标本中的白血病相关融合基因的RNA逆转录后,对相应的cDNA进行定性检测,以确定其融合基因的类型,从而及时辅助白血病临床诊断、临床药物的选择及评估疾病预后提供重要信息。本试剂盒检测结果仅供临床参考,不得作为临床诊断的唯一标准。建议结合患者临床表现和其他实验室检测对病情进行综合分析。
(三)产品包装规格
20测试/盒
(四)产品检验原理
本产品由白血病相应融合基因特异性引物、荧光探针、RT酶及Taq酶等成分组成,采用核酸扩增技术结合荧光标记探针杂交方法,通过荧光信号的变化,利用荧光标记定性检测人骨髓标本中的白血病相关融合基因RNA的转录产物cDNA。本试剂盒还使用尿苷酶(UNG)防污染体系,经加热可选择性地降解U-DNA,以防止先前PCR扩增产物的污染;采用内参基因(ABL1),控制整个试剂盒检测过程的有效性。
二、临床前研究概述
(一)主要原材料
1.主要原材料的选择
本试剂盒主要原材料包括:本产品在制备过程中主要原材料包括阳性对照原液、引物和探针、Taq酶、UNG酶、RT酶和RNasin酶。其中引物、探针为申请人自行设计后由专业的合成公司合成,阳性对照原液为企业自制,其他原材料均为外购方式获得。申请人选择有资质的供应商提供的原料,通过功能性试验,筛选出最佳原材料和供应商,并制定了各主要原材料的质量标准并经检验合格。
2.企业参考品设置情况
申请人设计了完整的企业参考品,包括准确性参考品、分析特异性参考品、检测限参考品和精密度参考品。其中:
准确性参考品共28份,包括检定合格的相应白血病融合基因阳性假病毒或内参基因假病毒,RNA浓度为3×104copies/μL。
分析特异性参考品共3份,包括骨髓细胞裂解物、HL60细胞株裂解物、Jurkat细胞株裂解物各1份,抽提为RNA,RNA浓度为3×104copies/μL。
检测限参考品共18份,包括检定合格的相应白血病融合基因阳性假病毒或内参基因假病毒,抽提为RNA后配制,终浓度为每种RNA 800copies/μL。
精密度参考品共10份,包括检定合格的相应白血病融合基因阳性假病毒或内参基因假病毒,抽提为RNA后配制,高浓度精密度的终浓度为每种RNA 2×104copies/μL,低浓度精密度的终浓度为每种RNA 2×103copies/μL。
(二)生产工艺及反应体系研究
申请人通过对试剂主要生产工艺的研究,确定了最佳生产工艺。
申请人对反应体系中的引物、探针的组合;引物浓度、探针浓度;缓冲液的选择;Taq酶、UNG酶用量;退火温度,退火时间,反应循环数,变性温度和时间;样本的上样量,抽提方法的选择;样本提取RNA质量要求;融合基因组合检测的设置等进行筛选和优化。通过功能性试验,最终确定了最佳反应体系。
(三)分析性能评估
分析性能包括核酸提取纯化、准确性、精密度、最低检出限、分析特异性(交叉反应和干扰试验)的评估等。申请人提交了有效运行的质量管理体系下生产的三批产品在适用机型上的性能评估资料。
在核酸提取纯化研究中,申请人评价了抽提方法的提取效率、提取RNA浓度的重复性、RNA纯度和抗干扰能力,确定了一种抽提试剂盒与该产品配套使用。
在准确性研究中,申请人检测企业准确性参考品和64例不同融合类型的阳性临床样本结果显示:阳性符合率均为100%。
在分析特异性研究中,申请人检测包括3份企业特异性参考品样本和8例融合基因阴性的临床样本,其阴性符合率为100%。
试剂盒检测6例SET-NUP214、NUP98-HOXA9、TLS-ERG融合基因阳性白血病患者骨髓血样本提取的核酸,其阴性符合率为100%。申请人选择4例融合基因阴性骨髓样本,通过添加的形式,分别研究HBV病毒、CMV病毒、HCV病毒和EBV病毒对检测结果的影响,试验结果显示试剂盒不受上述病原体的影响。干扰试验研究结果表明,轻度溶血的全血样本(即Hb浓度≤3g/L)、重度脂血的全血样本(TG浓度为20mmol/L)、重度黄疸的全血样本(T-Bil含量为172umol/l)对检测结果无干扰。2.5倍治疗水平浓度的常见药物和治疗白血病的靶向药物,阿莫西林,阿司匹林、维甲酸、伊马替尼、阿糖胞苷、长春新碱、柔红霉素、亚砷酸、左旋门冬酰胺酶、环磷酰胺和氟达拉滨研究结果显示药物对检测结果无影响。
在精密度研究中,申请人采用三批试剂盒,对各精密度参考品,进行批次内/间、试验日内/间、试验轮次内/间、操作者间、实验室间精密度研究,CV值≤5%。试剂盒对18例临床样本进行三批产品的精密度试验,均能检出,且其实验数据CV值≤5%。对15例临床样本进行20天精密度试验,从批次内、批次间、不同仪器、试验操作者间、实验室间精密度进行研究,检测结果均能检出,且其实验数据CV值≤5%。
在最低检出限研究中,申请人采用相应白血病融合基因阳性样本或内参基因假病毒分别稀释至10000copies/反应、1000copies/反应、100copies/反应确定试剂盒检测限。申请人采用覆盖声称靶基因类型的阳性标本与阴性样本稀释至检测限附近进行最低检出限的验证,结果显示所有样本重复20次,均≥95%检出。
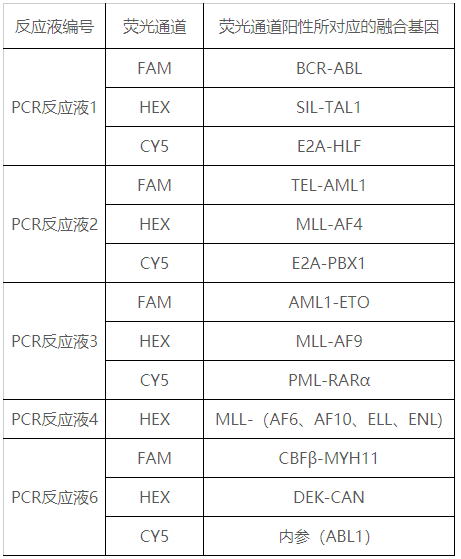
(四)阳性判断值
申请人采用ROC曲线法确定阳性判断值。申请人采用69个不同融合基因型阳性样本和47个阴性样本进行检测,建立检测试剂的阳性判断值,申请人选取18例白血病相应15种融合基因阳性和10例相应15种融合基因阴性共28例临床诊断结果明确的标本来验证试剂盒的准确性。检测结果显示建立的阳性判断值可以满足试剂盒对于临床样本准确性的要求。
申请人选取125例临床样本,包含覆盖声称靶基因阳性样本、检测融合基因范围外的融合类型阳性样本和融合基因阴性样本。对建立的阳性判断值进行验证。检测结果显示建立的阳性判断值可以满足试剂盒对于临床样本的检测。最终确定以下判读方法:
因不同反应液、不同荧光通道检测不同的融合基因,因此,每个样本在分析中,必须逐一对不同的反应液、同一反应液中的不同通道分别分析,即必须逐一将样本、阳性对照、阴性对照同时分析其在各个反应液中的三种荧光扩增曲线。
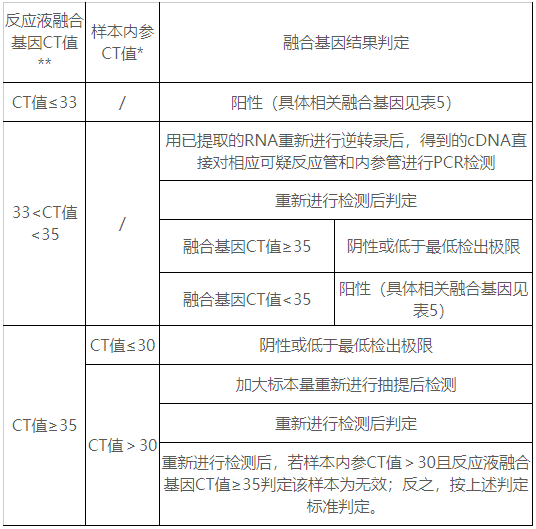
表3相关融合基因对应表
(五)稳定性研究
申请人对本产品的实时稳定性、冻融稳定性等进行了研究,确定了在各种条件下本产品的有效保存时间。同时,分别对骨髓样本和抽提后的RNA保存条件、冻融时间及冻融次数进行了研究确定了临床样本的保存条件。
实时稳定性研究:将三批试剂盒在-20℃±4℃冰箱中储存0、10、15、18个月后分别进行一次成品检验,结果表明试剂盒在常规储存条件下18个月后检测结果均符合技术要求,故本试剂盒常规稳定性定为:储存条件-20℃±4℃,储存18个月。
冻融次数研究:使用一批试剂盒进行开盖试验,具体操作流程如下:在-20℃±4℃冰箱中存放的试剂盒2盒,每5天左右拿出一次,将试剂完全融解后,将试剂完全融解,开盖2分钟后再盖好盖子,放回-20℃±4℃冰箱中存放,每5天左右重复一次,连续5次,1个月后进行检定,试剂盒准确性、特异性、检测限性能稳定,因此将试剂盒开盖、反复冻融条件定为:有效期内反复冻融4次,性能稳定。
三、临床评价概述
申请人在苏州大学附属第一医院、浙江大学医学院附属第一医院、上海交通大学医学院附属上海儿童医学中心和首都医科大学附属北京儿童医院共四家临床试验机构完成了临床试验。采用试验体外诊断试剂与染色体核型分析、Sanger测序法以及已上市同类产品分别进行比较研究,对产品临床性能进行评价。入组病例主要包括急性髓系白血病、急性淋巴细胞白血病和慢性粒细胞白血病等血液病患者,样本类型为骨髓样本。临床试验共入组受试者样本1103例,包括74例核型分析失败患者样本,其中融合基因阳性样本639例,阴性样本464例;融合基因阳性样本中包括BCR-ABL阳性172例、AML1-ETO阳性100例、PML-RARα阳性100例、E2A-HLF阳性9例、MLL-AF4阳性30例、SIL-TAL1阳性16例、E2A-PBX1阳性22例、TEL-AML1阳性50例、CBFβ-MYH11阳性40例、DEK-CAN阳性16例、MLL-AF6、MLL-AF10、MLL-ELL、MLL-ENL联合检测阳性61例、MLL-AF9阳性23例。
经统计核验,试验体外诊断试剂与染色体核型分析对比,临床灵敏度为99.76%(95%CI:98.63%,99.96%),临床特异度为74.56%(95%CI:70.99%,77.82%),不一致样本采用Sanger测序法复核,并结合临床分子分型结果进行分析,测序结果以及临床分子分型结果与试验体外诊断试剂检测结果100%一致。分析原因,不一致结果主要是因为染色体核型分析检测融合基因灵敏度偏低导致。74例核型分析失败样本检测结果,试验体外诊断试剂与Sanger测序法检测结果符合率100%。
试验体外诊断试剂与Sanger测序法对比试验结果显示,两种方法针对每一种被测融合基因检测阳性符合率、阴性符合率均为100%;收集临床机构针对受试者的分子分型诊断结果,试验体外诊断试剂针对被测融合基因检测结果与临床机构分子分型诊断结果100%一致。
试验体外诊断试剂与BCR-ABL、AML1-ETO、PML-RARα已上市同类产品对比试验分别检测临床样本200例,结果显示,BCR-ABL阳性符合率100%(95%CI:90.36%,100%),阴性符合率100%(95%CI:97.71%,100%);AML1-ETO阳性符合率100%(95%CI:92.44%,100%),阴性符合率100%(95%CI:97.55%,100%);PML-RARα阳性符合率100%(95%CI:91.24%,100%),阴性符合率100%(95%CI:97.66%,100%)。
综上所述,该产品临床试验设计符合《体外诊断试剂临床试验技术指导原则》的相关要求。临床试验结果显示该产品临床灵敏度、临床特异度满足临床要求。
四、产品受益风险判定
根据申请人提供的申报资料,经综合评价,在目前认知水平上,认为该产品的上市为适用人群带来的受益大于风险。但为保证用械安全,基于对主要剩余风险的规避,需在说明书中提示以下信息:
本试剂盒检测结果仅供临床参考,不得作为临床诊断的唯一标准。建议结合患者临床表现和其他实验室检测对病情进行综合分析。
本申报项目为境内第三类医疗器械产品注册,属于境内同品种首个产品。申请人的注册申报资料符合现行要求,依据《医疗器械监督管理条例》(国务院令第680号)、《体外诊断试剂注册管理办法》(国家食品药品监督管理总局令2014年第5号)等相关医疗器械法规与配套规章,经系统评价后,建议准予注册。
2022年2月24日
附件:产品说明书

