受理号:CSZ1900370
体外诊断试剂产品注册技术审评报告
产品中文名称:SHOX2/RASSF1A/PTGER4 基因甲基化检测试剂盒(PCR-荧光探针法)
产品管理类别:第三类
申请人名 称:北京艾克伦医疗科技有限公司
国家药品监督管理局
医疗器械技术审评中心
基本信息
一、申请人名称 北京艾克伦医疗科技有限公司
二、申请人住所 北京市昌平区科技园区白浮泉路10号2号楼北控科技大厦3层302室
三、生产地址 北京市昌平区科技园区白浮泉路10号2号楼北控科技大厦3层302室、北京市昌平区超前路37号1幢4层5-4-2-01
技术审评概述
一、产品概述
(一)产品主要组成成分 产品主要组成成分见下表:
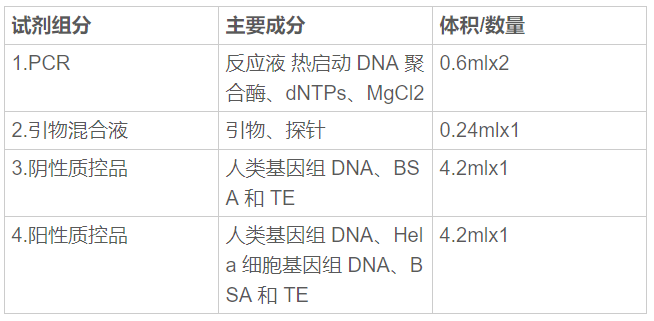
(二)产品预期用途
本试剂盒用于体外检测人外周血血浆中SHOX2、RASSF1A、PTGER4基因的甲基化。
本产品用于临床上对疑似肺癌患者的辅助诊断,检测结果阳性不作为肺癌早期诊断或确诊的证据,检测结果阴性也不能排除肺癌的可能。该检测不能作为肿瘤早期诊断或确诊的依据,不宜用于普通人群的肿瘤筛查。SHOX2 属于 SHOX 基因家族,在胚胎形成期对骨骼、心脏和神经系统的发育作用重大,在肺癌、乳腺癌和肾癌中异常表达。RASSF1A 调控涉及基因转录、信号转导、细胞周期、细胞凋亡等多种生物学功能,可以通过多种途径抑制肿瘤形成。PTGER4 属于 G 蛋白偶联受体家族,是非常重要的抑癌基因。研究发现,肺癌患者血浆样本中 SHOX2、RASSF1A 及 PTGER4三基因启动子区域呈高度甲基化。
(三)产品包装规格
30 人份/盒。
(四)产品检验原理
提取血浆中的游离 DNA,然后用亚硫酸盐转化未发生甲基化的胞嘧啶,通过脱氨基反应产生尿嘧啶磺酸盐,发生甲基化的胞嘧啶则不会被亚硫酸盐转化。将亚硫酸盐转化的 DNA(BisDNA)做多重 PCR 扩增,PCR 反应中的引物、探针能区分甲基化和非甲基化序列,甲基化序列优先得到扩增,与甲基化 SHOX2、RASSF1A、PTGER4 基因序列特异性结合的荧光素探针可以在 PCR 反应中专一地检测出甲基化序列。内参对照ACTB(β-actin)基因用于评估检测中 DNA 量是否足够。
二、临床前研究概述
(一)主要原材料
1.主要原材料的选择
本产品的主要原材料包括引物干粉、PCR反应液、人类基因组DNA和Hela细胞基因组DNA。这些原料均是通过外购的方式获得。
其中引物的序列均由申请人自行设计,由合成公司经过合成、修饰、纯化方式获得;PCR反应液由供应商化学合成获得;人类基因组DNA和Hela细胞基因组DNA由供应商提取纯化获得。
申请人对主要原材料进行了供应商的选择,通过功能性实验筛选出合格供应商,制定了各主要原材料的技术要求和质量标准并经检验合格。
2.企业参考品设置情况
本产品企业参考品包括阳性参考品、阴性参考品、精密度参考品以及检测限参考品,组成如下:
阳性参考品包括 12种。P1、P2、P3为一定 DNA 浓度下SHOX2、RASSF1A、PTGER4 三基因均为甲基化的不同甲基化比例的参考品;SP1、SP2、SP3为一定 DNA 浓度不同比例的SHOX2单甲基化参考品;RP1、RP2、RP3为一定 DNA 浓度不同比例的RASSF1A单甲基化参考品;PP1、PP2和PP3为一定DNA 浓度不同比例的PTGER4单甲基化参考品。阴性参考品包括 4种,包括三种不同DNA浓度下SHOX2、RASSF1A、PTGER4均无甲基化的参考品,一种三基因均甲基化但甲基化比例低于检测限的阴性参考品。精密度参考品包括6种。J1、J2为一定DNA 浓度,SHOX2、RASSF1A、PTGER4 三基因均甲基化,且甲基化程度高低不同的两种精密度参考品。SJ1、SJ2为一定DNA 浓度,SHOX2单基因甲基化,且甲基化程度高低不同的两种精密度参考品。RJ1、RJ2为一定DNA 浓度,RASSF1A单基因甲基化,且甲基化程度高低不同的两种精密度参考品。PJ1、PJ2 为一定DNA 浓度,PTGER4基因单甲基化,且甲基化程度高低不同的两种精密度参考品。
检测限参考品包括 4 种。包括低 DNA 浓度的 SHOX2、RASSF1A、PTGER4三基因甲基化参考品,低 DNA 浓度的SHOX2单基因甲基化参考品,低 DNA 浓度的RASSF1A单基因甲基化参考品,低 DNA 浓度的 PTGER4单基因甲基化参考品。
(二)生产工艺及反应体系研究
申请人对试剂盒反应体系的研究中包括引物探针浓度的确定、PCR 反应液的选择、阴/阳性质控品配方的确定等;对PCR过程中的退火温度进行研究;完成样本的用量以及样本保存时间 的 研 究 ;对 该 产 品 三 种 适 用 机 型 宏 石 PCR 分 析 系 统SLAN-96P、Applied Biosystems 7500,Roche Lightcycler 480的反应程序及分析条件进行了研究。
通过功能性实验,最终确定了最佳的反应体系。申请人根据试剂盒中试剂及组件的主要生产工艺的研究结果,确定了最佳的生产工艺。
(三)分析性能评估
本产品分析性能评估内容包括:准确性(阳性符合率、阴性符合率)、精密度、检测限、分析特异性的评估;核酸提取试剂的性能评估;三种适用的 PCR 仪机型(宏石 PCR 分析系统SLAN-96P、Applied Biosystems 7500,Roche Lightcycler 480)评估。
准确性研究使用企业阴/阳性参考品及临床样本来源的阴性/阳性/弱阳性样本池分别对三个批次的试剂盒进行检测,检测结果显示阳性符合率 100%、阴性符合率 100%。同时用试剂盒与同类对比试剂分别检测临床样本,包括各期肺癌样本共 128 例及肺结核样本 32 例,结果显示试剂盒对于Ⅰ~Ⅳ期肺癌和肺结核样本监测的准确率分别为 78.13%、87.50%、84.38%、87.50%和81.25%。
精密度的研究使用了两种不同的样本,第一种样本是精密度参考品,对三批次试剂盒分别在三种适用的仪器上进行检测。
检测结果显示试剂盒检测的 SHOX2、RASSF1A、PTGER4 和ACTB 基因的变异系数均小于 10%。第二种样本是通过在临床正常人血浆样本中添加一定浓度的细胞系 DNA 模拟精密度企业参考品进行验证,检测结果显示 SHOX2、RASSF1A、PTGER4和 ACTB 基因的变异系数均小于 10%。同时也对配套用核酸提取试剂的精密度进行了研究,结果表明在样本采集过程、核酸提取过程中 DNA 提取量变异系数小于 10%;核酸转化过程中,ACTB 基因 Ct 值的变异系数均小于 10%。检测限研究,以阴性血浆样本为基质,对不同游离 DNA 浓度下含有不同比例甲基化 DNA 的血浆样本进行检测,得到血浆样本的检测限,并在三种适用机型上进行验证。结果表明试剂盒可检测血浆中阳性 DNA 最低浓度为 0.1pg/?L。在血浆 DNA浓度为 0.02ng/?L 时,产品检测 P 值为阳性的检测限为 0.5%三基因甲基化,单基因甲基化的检测限分别为 0.2%的 SHOX2 甲基化,0.5%的 RASSF1A 甲基化和 0.2%的 PTGER4 甲基化。使用三批试剂盒对四种检测限参考品进行检测验证,结果表明对于DNA浓度为0.02ng/?L的参考品,P值为阳性的检测限为0.5%的三基因甲基化,单基因甲基化的检测限分别为 0.2%的 SHOX2甲基化,0.5%的 RASSF1A 甲基化和 0.2%的 PTGER4 甲基化。特异性研究,交叉反应实验结果表明试剂盒检测干扰样本的综合特异性超过 85%。对于发生肺转移的其它原发癌症(包括肝癌、胃癌、胰腺癌、乳腺癌、食管癌、结直肠癌)检出率较低,假阳性率低于 25%,说明产品仅适用于原发性肺癌的检测。检测自身免疫性疾病包括红斑狼疮和类风湿性关节炎的特异性超过 90%,假阳性率低于 10%。检测其它肺部疾病的特异性超过 85%,假阳性率低于 15%。检测宫颈癌特异性为 80%,假阳性率为 20%。
干扰实验显示,样本中含有以下干扰物:未甲基化 DNA(150ng/ml)、胆红素(0.20mg/ml)、血红蛋白(10mg/ml)、甘油三酯(12mg/ml)、蛋白(血清白蛋白,120mg/ml)、红细胞(0.4% v/v)、K2EDTA(20mg/ml)、胆固醇(5mg/ml)、尿酸(0.235mg/ml)和葡萄糖(10mg/ml),对检测结果无影响。常见治疗药物,包括感冒药、消炎药、心脑血管疾病药、糖尿病药、胃病药、维生素、高血压药、安定药、镇痛药等在最大使用剂量范围内,不影响试剂盒对样本检测的 Ct 值及 P 值判定。
对与试剂盒联合使用并在说明书中推荐的血浆样本处理试剂(核酸提取试剂,京昌械备 20180004 号,北京艾克伦医疗科技有限公司)进行了性能评估,结果表明产品配套使用的核酸提取试剂,提取效率、保护效率和转化效率分别为 85.18%、93.52%和 99.32%。
对该产品适用的三种仪器宏石 PCR 分析系统 SLAN-96P、Applied Biosystems 7500,Roche Lightcycler 480 进行了性能评估,结果显示在三种适用机型上的阴/阳性符合率、检测限、精密度均符合质量要求,可以作为该产品的检测 PCR 仪。
(四)阳性判断值研究
申请人采用三基因拟合 P 值公式法确定阳性判断值。每个样本进行 3 次 PCR 平行测试,计算 SHOX2、RASSF1A、PTGER4和 ACTB 基因 3 个复孔平均 Ct 值,其中各个基因无扩增时,Ct值定义为 45.0。ACTB 基因的 Ct 值≤35.0 时,样本检测结果有效。在确定样本检测结果有效的情况下,样本 P 值≥2.0 则判断样本为阳性,样本 P 值<2.0 则判断样本为阴性。
阳性判断值研究入组(训练集和验证集)有效样本数共计489 例,包括肺部正常样本 207 例、肺癌病人样本 141 例、其它干扰样本(包括结节、肺炎、肺结核、囊肿、食管癌、胃癌等)141 例。样本数据进行多种方法统计分析,包括 1/3 次 PCR 反应 Ct 值、2/3 次 PCR 反应 Ct 值、3/3 次 PCR 反应 Ct 值、3 次PCR 反应平均 Ct 值、单基因、两基因、三基因、三基因组合拟合公式等,比较不同分析方法的检测灵敏度、特异性、总符合率,最终确定使用三基因拟合 P 值公式法检测准确率最高。并使用测试集样本再次验证三基因拟合公式和其他阳性判断值方法,测试集研究入组有效例数 248 例,包括肺部正常人样本 81例,肺癌病人样本 76 例,其他干扰样本 91 例。结果表明三基因拟合 P 值公式法特异性为 94.19%(95%CI:90.65%-97.72%),灵敏度为 89.47%(95%CI:82.41%-96.53%),总符合率为 92.74%(95%CI:89.49%-95.99%)。
(五)稳定性研究
申请人对该产品的稳定性的研究包括长期稳定性、使用稳定性(包括开瓶稳定性、冻融稳定性和热稳定性)及样本稳定性(包括全血、血浆及 BisDNA 的稳定性)。长期稳定性:选择三批次试剂盒置于-20±5℃冰箱中保存,在储存后的第 0、3、6、9、12、13、14、15 个月进行检测,结果显示产品在生产后 15 个月的产品性能符合技术要求,试剂性能稳定。确定产品有效期可达 12 个月。
使用稳定性:将三批试剂盒置于-20±5℃开瓶放置 6 周后,稳定性结果 P 值及 CV 值均无显著差异,无显著变化趋势,其产品的主要性能均符合技术要求。将三批次试剂盒在规定的储存条件下,取出试剂盒反复冻融 6 次,检测每次冻融后的试剂盒的性能均符合技术要求。将三批试剂盒置于 37℃,分别于 1天、2 天、3 天后检测产品性能,均符合技术要求。申请人对全血样本、血浆样本及 BisDNA 的样本稳定性进行了研究,研究结果表明使用 EDTA 抗凝管(BDVacutainer?,6mL,国械注进 20152222083)采集全血,建议选用 2-8℃放置4 小时以内的新鲜血样分离血浆后用于检测;使用康为游离DNA 采血管(康为世纪,5mL,苏械注准 20192220059)采集全血,建议选用室温放置 4 天以内的血样分离血浆后用于检测。分离后的血浆样本建议在-20±5℃保存 30 天以内,在 2-8℃保存12 小时以内进行检测;BisDNA 保存期限为在-20±5℃保存 4 天以内,在 2~8℃保存 16 小时以内进行 PCR 检测。
三、临床评价概述
本产品在上海市东方医院、山西省肿瘤医院和河南省肿瘤医院三家临床试验机构进行临床试验,采用试验体外诊断试剂与临床参考标准进行比较研究,确认本产品的临床性能。其中,肺癌和其他肿瘤病例采用病理诊断确诊,其他疾病根据相关诊疗指南采用病理诊断或 CT 等综合诊断确诊。入组病例为肺癌疑似病例,样本类型为血浆。产品临床灵敏度和特异度评价共纳入临床病例 1303 例,其中肺癌病例 486 例(覆盖肺癌所有分期及所有病理分型),非肺癌的其他病例 817 例(包括其他易产生干扰的肿瘤及各种良性疾病病例)。试验结果显示:本产品临床灵敏度为 86.83%(95%CI:83.53%~89.55%),特异度 95.59%(95%CI:93.96%~96.80%),总符合率为 92.33% (95%CI:90.75%~93.65%) 。上述结果显示试验体外诊断试剂具有较好的临床灵敏度和特异度,满足临床使用需求。
此外,临床试验还纳入 260 例肺癌疑似病例(包括本产品与临床参考标准检测结果不符样本),采用试验体外诊断试剂与一代测序进行比较研究,确认本产品的临床检测性能。试验结
果显示:针对 SHOX2 基因,阳性符合率为 100%(95%CI:97.37%~100%),阴性符合率为 98.31%(95%CI:94.03%~99.53%),总符合率为 99.23%(95%CI:97.24%~99.79%);针对 RASSF1A 基因,阳性符合率为 100%(95%CI:97.24%~100%),阴性符合率为 98.28%(95%CI:93.93%~99.53%),总符合率为 99.23%(95%CI:97.24%~99.79%);针对 PTGER4基因,阳性符合率为 100%(95%CI:96.97%~100%),阴性符合率为 98.54%(95%CI:94.83%~99.60%),总符合率为 99.23%(95%CI:97.24%~99.79%)。上述结果显示两者之间具有良好的一致性,本产品临床检测性能满足要求。
另外,采用试验体外诊断试剂对 37 例肺癌患者手术前后的样本进行连续检测,35 例样本术前检测结果为阳性,术后检测结果为阴性;2 例样本术前术后检测结果均为阴性,表明肺癌手术切除后血浆中甲基化水平降低。
综上所述,临床试验结果显示本产品的临床性能满足技术审评要求。
四、产品受益风险判定
根据申请人提供的申报资料,经综合评价,在目前认知水平上,认为该产品上市带来的受益大于风险。但为保证用械安全,基于对主要剩余风险的规避,需要在说明书中提示以下信息:
1.使用普通 EDTA 真空采血管采集后的血样应立即分离血浆,若不能立即分离血浆,应在 2~8℃保存,保存时间不超过 4小时;使用游离 DNA 采血管采集后的血浆可以室温保存 4 天。不得冰冻血样。
2.禁止使用离心机刹车(急停)功能,以防止破坏血细胞层。
3.制备好的血浆样本可以在-20±5℃下保存不超过 30 天可以在 2~8℃存放不超过 12 小时。
4.BisDNA 不立即使用,可在 2~8℃保存 16 小时,或在-20±5℃保存 4 天。
5.PCR 反应液和引物混合液使用完毕立即复冻。
6.密封后的 PCR 管可在 2~8℃放置不超过 2 小时。
7.只能用于体外诊断。
8.该产品的有效性仅针对于 EDTA 抗凝采血管。对于其它血样的收集方式有效性没有做测试。
9.该产品的使用者应该是接受过 PCR 反应训练的试验者。
10.由于肺癌检测依赖于样品中肿瘤 DNA 的量,所以可能受样品收集过程、样品储存方式、病人个体因素(如年龄,其它疾病)以及肿瘤级别影响,外周血的采集、血浆的制备和存储均应按照要求进行,否则将影响检测结果,导致假阴性的检测结果。
11.由于血浆样本中游离 DNA 含量低、易降解,应严格按照【样本要求】的规定处理和保存样本,否则影响检测,造成假阴性结果。
12.由于扩增的初始靶 DNA 含量极低,故扩增循环数较长。应尽量避免检测环境的污染,否则容易在扩增循环末期产生假阳性信号。
综合评价意见
本申报项目为境内第三类体外诊断试剂产品注册,属于境内同品种首个产品。申请人的注册申报资料符合现行要求,依据《医疗器械监督管理条例》(国务院令第 680 号)、《体外诊断试剂注册管理办法》(国家食品药品监督管理总局令 2014 年第 5号)等相关医疗器械法规与配套规章,经系统评价后,建议准予注册。
2022 年 1 月 29 日





