体外诊断试剂产品注册技术审评报告
产品中文名称:结核分枝杆菌复合群核酸检测试剂盒(PCR-荧光探针法)
北京市北京经济技术开发区康定街1号14号楼3层1室
北京市北京经济技术开发区康定街1号14号楼2层、北京市北京经济技术开发区康定街1号10号楼2层
技术审评概述
一、产品概述
(一)产品主要组成成分
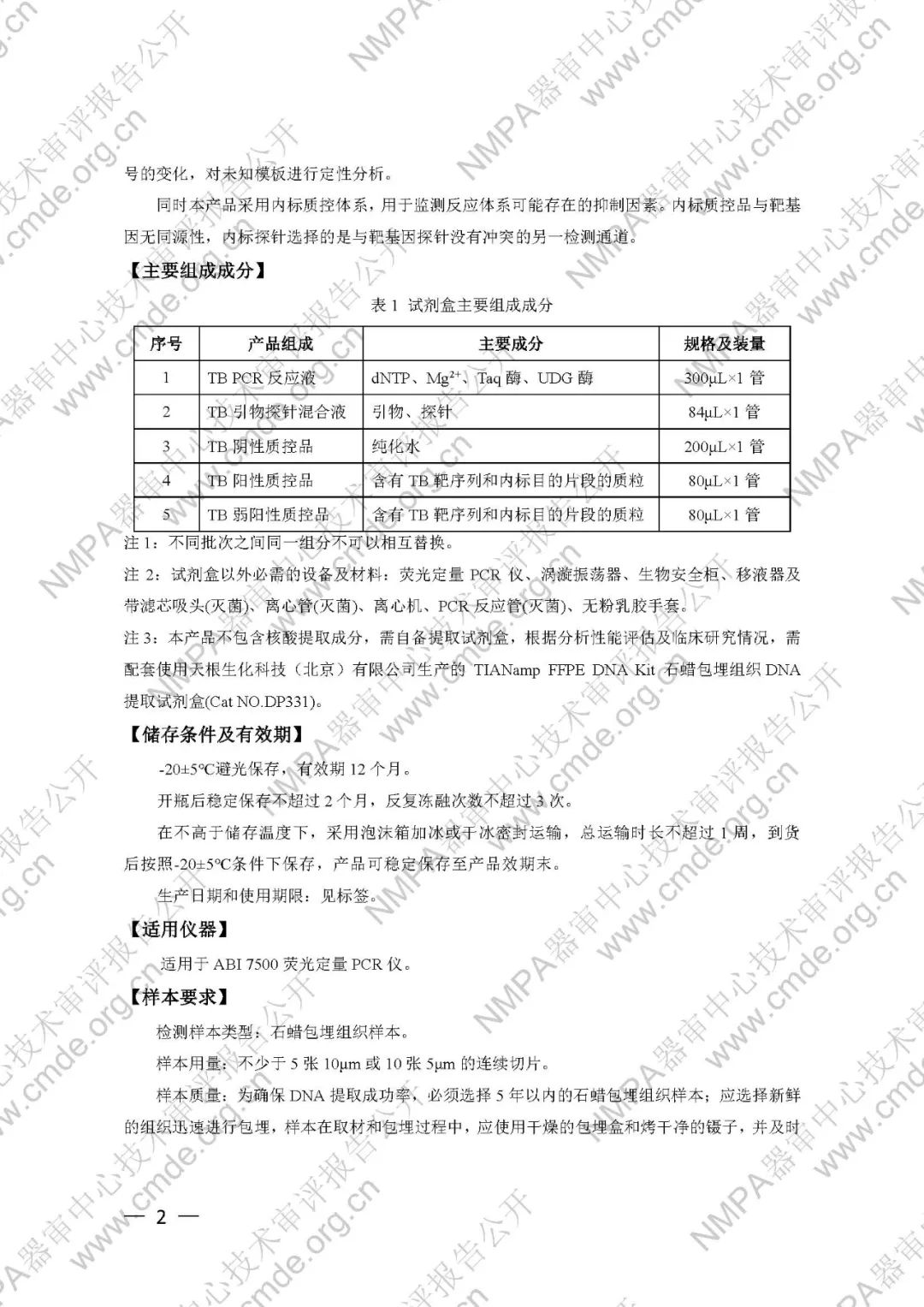
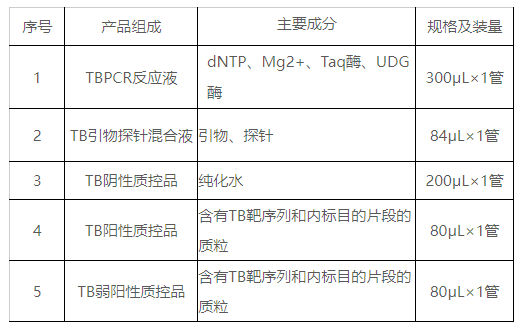
本试剂盒含有TBPCR反应液、TB引物探针混合液、TB阴性质控品、TB阳性质控品、TB弱阳性质控品,主要组成成分见表1。
表1试剂盒主要组成成分
本试剂盒用于体外定性检测人福尔马林固定、石蜡包埋组织样本中的结核分枝杆菌复合群核酸。
本产品采用PCR荧光探针技术,用于辅助结核病的病理诊断。
结核分枝杆菌(tuberclebacilli,TB)复合群主要包括结核分枝杆菌、牛结核分枝杆菌、非洲分枝杆菌和田鼠分枝杆菌,除田鼠分枝杆菌外,其余三种均对人致病,是结核病的病原菌,可通过呼吸道、消化道和破损的皮肤粘膜进入机体,侵犯多种组织器官,引起相应器官的结核病,以肺结核最常见。其致病作用可能与细菌在组织细胞内顽强增殖引起炎症反应,以及诱导机体产生迟发型变态反应性损伤有关。TB基因组中的插入序列IS6110具有特异性强,重复性好的特点,是TB检测常用的靶基因片段。
实验操作人员应接受过基因扩增或分子生物学方法检测的专业培训,具备相关的实验操作资格,实验室应具备相关的生物安全防备设施及防护程序。
该产品检测结果应结合患者临床症状、体征、流行病学背景及其他临床诊断结果进行综合判断,不得作为疾病诊断的唯一标准。
本产品检测的靶基因为TB基因组中的插入序列IS6110,该序列作为靶基因具有特异性强,重复性好等特性。内标为Her2基因,在人基因组中表达稳定,是常见的管家基因。本产品采用荧光PCR技术,选取结核分枝杆菌基因组中相对保守的区域,设计特异性引物及探针,在样本提取之后采用荧光PCR对TBDNA进行快速检测。
本产品采用了Taqman荧光探针技术,其试剂比常规PCR试剂多了一个寡聚核苷酸探针,这个探针带有一个荧光发光基团和一个荧光淬灭基团,完整的探针在特定光源激发下,发光基团所产生的荧光被淬灭基团全部吸收,样品无荧光。PCR过程中,Taq酶在延伸DNA链的同时,可通过自身的5’→3’核酸外切酶活性降解与模板结合的特异性荧光探针,使荧光报告基团与淬灭基团分离,分离后的荧光报告基团在特定光源激发下产生荧光。通过监测整个PCR过程荧光信号的变化,对未知模板进行定性分析。
同时本产品采用内标质控体系,用于监测反应体系可能存在的抑制因素。内标质控品与靶基因无同源性,内标探针选择的是与靶基因探针没有冲突的另一检测通道。
本试剂盒主要原材料包括:引物、探针、qPCRMasterMix、UDG酶、dUTP、TB菌液。其中引物、探针为申请人自行设计后由专业的合成公司合成,其他原材料均为外购方式获得。申请人选择有资质的供应商提供的原料,通过功能性试验,筛选出最佳原材料和供应商,并制定了各主要原材料的质量标准并经检验合格。
企业参考品设置情况:申请人设计了完整的企业参考品,包括阳性参考品、阴性参考品、最低检出限参考品和精密度参考品。其中:
阳性参考品共15份,包括肺结核样本、右侧颈结核样本、淋巴结核样本、非洲分枝杆菌样本、骨结核样本、左肩结核样本、肾结核样本、腰椎结核样本、左胸结核样本、直肠结核样本、胸壁结核样本、BCG菌株和结核分枝杆菌菌株,浓度在1×103copies/mL~5×105copies/mL之间。
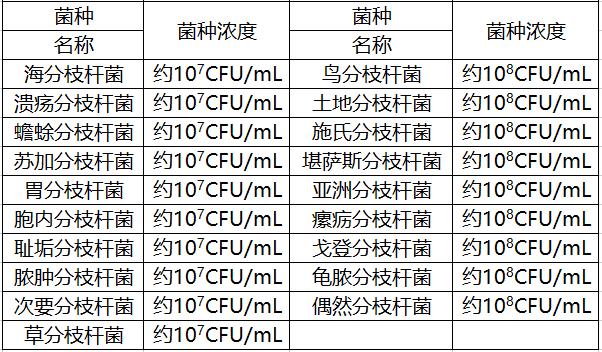
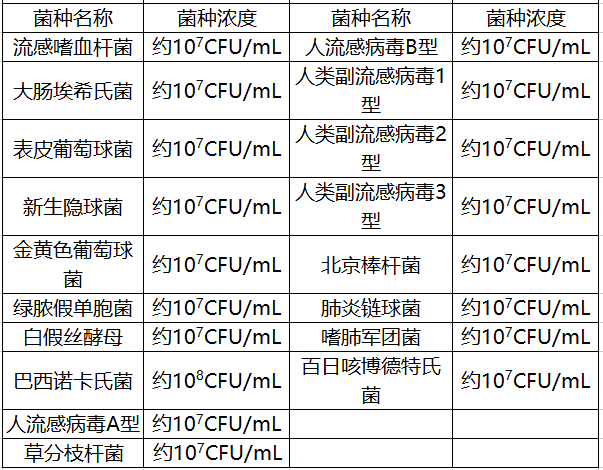
阴性参考品共15份,包括浸润性腺癌样本、肺癌样本、慢性肺部炎症样本、间质性肺炎样本、肺鳞状细胞癌样本、人流感病毒A型菌株、人流感病毒B型菌株、脓肿分枝杆菌、蟾蜍分枝杆菌、鸟分枝杆菌、苏加分枝杆菌和海分枝杆菌,其中菌株的浓度均为106CFU/mL。
最低检出限参考品共8份,包括肺结核样本、非洲分枝杆菌样本、淋巴结核样本、骨结核样本、右颈结核样本、腰椎结核样本、BCG菌株和结核分枝杆菌菌株,浓度均为1×103copies/mL。
精密度参考品共2份,均为肺结核样本,浓度分别为1×106copies/mL和1×104copies/mL。
申请人通过对试剂主要生产工艺的研究,确定了最佳生产工艺。
申请人对反应体系中的TBPCR反应液、引物探针序列、引物探针浓度、样本的上样量、内标体系和UDG酶体系的干扰、退火温度、反应循环数等进行筛选和优化,通过功能性试验,最终确定了最佳反应体系。
该产品的分析性能包括核酸提取纯化、准确性、精密度、最低检出限、分析特异性(交叉反应和干扰试验)、包容性、不同部位组织的评估等。申请人提交了有效运行的质量管理体系下生产的三批产品在适用机型上的性能评估资料。
在核酸提取纯化研究中,申请人采用临床石蜡包埋组织样本,平行比较了2种石蜡包埋组织核酸提取试剂盒的提取效果,根据与该产品的组合性能研究,确定了1种核酸提取试剂盒,与该产品配套使用。
在准确性研究中,申请人采用三批试剂盒检测15份企业阳性参考品(1×103copies/mL~5×105copies/mL)和15份企业阴性参考品(其中菌株浓度为106CFU/mL),结果显示:阳性符合率和阴性符合率均为100%。
在精密度研究中,申请人采用三批试剂盒,检测阴性样本(不含结核分枝杆菌复合群)、弱阳性样本(浓度为2×103copies/mL)和中等阳性样本(浓度为5×105copies/mL),分别评估了批内、批间、日间、不同操作者之间以及不同实验地点之间的精密度。结果显示:检测结果Ct值的CV值均小于5%,表明本产品的批内、批间、日间、不同操作者间以及不同实验地点间的重复性均符合要求。同时,对低浓度的石蜡包埋组织样本进行提取精密度检测,结果显示:提取后临床样本的DNA浓度及纯度差异较小,检测结果Ct值的CV值均小于5%,说明石蜡包埋组织样本经多次提取的DNA质量及检测结果一致性符合要求。
在最低检出限研究中,申请人将结核分枝杆菌、牛结核分枝杆菌石蜡样本分别与阴性石蜡样本混合,采用三批成品试剂盒检测,最终确定并验证了本试剂盒的最低检出限为1×103copies/mL。
在交叉反应研究中,申请人采用三批试剂盒,对易产生交叉反应的病原体与可能引发临床相似症状的病原菌进行了研究,包括19种非结核分枝杆菌复合群的其他分枝杆菌以及其他17种病原体。
本产品对以上36种病原菌的检测结果全部为阴性。
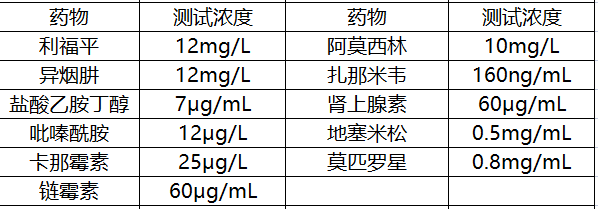
在干扰试验中,申请人对外源及内源干扰物质,肺癌样本、肺部良性疾病样本及肺部肉芽肿样本进行了研究。
结果显示:低于上述测试浓度的药物不会对本产品的检测结果产生干扰。
对其他外源性干扰物质10%福尔马林(0.05v/v)、95%乙醇(0.05v/v)进行研究,同时对内源性干扰物质血红素(0.02g/mL)、粘液(0.05v/v)、血红蛋白(0.2g/mL)、人基因组(2.5μg/mL)进行研究,结果显示:低于上述浓度的干扰物质不会对本产品的检测结果产生干扰。
采用肺癌样本、肺部良性疾病样本及肺部肉芽肿样本(共20余例)分别进行提取检测,结果显示:检测结果全部为阴性,以上样本类型不会干扰本试剂盒的检测能力。
在包容性研究中,申请人采用三批试剂盒,检测结核分枝杆菌、牛结核分枝杆菌、非洲分枝杆菌和田鼠分枝杆菌。结果显示:本产品对结核分枝杆菌(浓度为103copies/mL)、牛结核分枝杆菌(浓度为1×103copies/mL)、非洲分枝杆菌(浓度为2×103copies/mL)和田鼠分枝杆菌(浓度为2×103copies/mL)均能100%检出。
不同部位组织的评估:选择不同部位的结核病组织样本,包括睾丸结核、淋巴结核、肾结核、手指结核、胸壁结核、胸腔结核、胸椎结核、腰椎结核、右颈结核、右膝结核、直肠结核、左颈结核、左胸结核、左腰结核,提取核酸后,采用阴性样本稀释至最低检出限,用三批试剂分别对其检测20次,检测结果均为阳性,与测序结果一致。
(四)阳性判断值
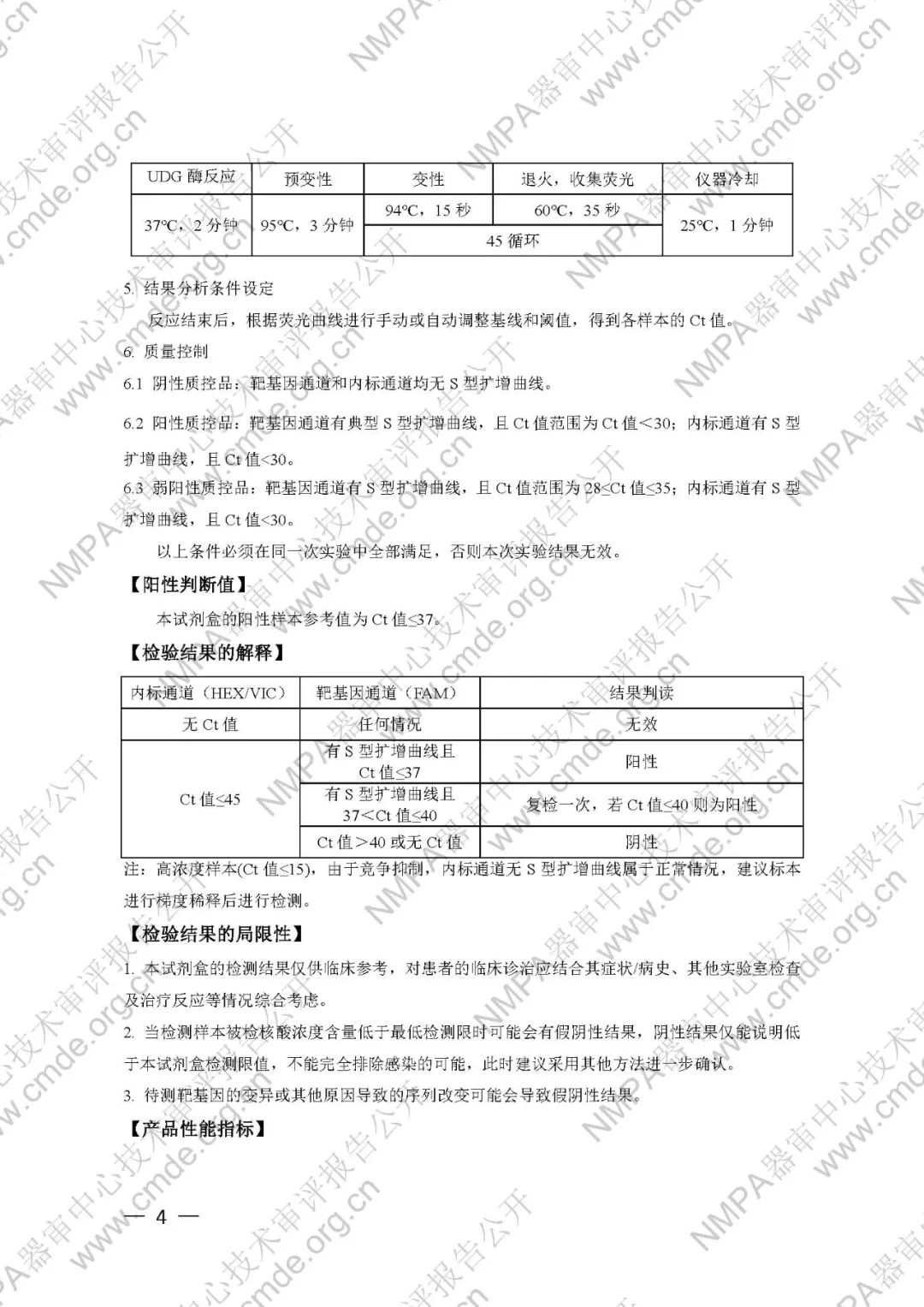
申请人采用ROC曲线法确定阳性判断值。申请人采用本产品对237例临床石蜡包埋组织样本(178个结核分枝杆菌复合群阳性样本和59个结核分枝杆菌复合群阴性样本)进行检测,结果显示:当靶基因通道Ct值为37左右时,本产品的灵敏度和特异性达到最佳。所以确定本产品的阳性判断值为37,并确定以下判读方法:

注:高浓度样本(Ct值≤15),由于竞争抑制,内标通道无S型扩增曲线属于正常情况,建议标本进行梯度稀释后进行检测。
申请人采用申报产品对17例灰区样本进行复检验证,结果显示:复检后的结果与测序验证的结果一致性可达到90%以上。
(五)稳定性研究
申请人对本产品的实时稳定性、运输稳定性、开瓶及冻融稳定性等进行了研究,确定了在各种条件下本产品的有效保存时间。同时,对石蜡包埋组织样本的常温放置稳定性、石蜡包埋组织提取的DNA冷冻稳定性进行了研究,确定了临床样本的有效保存时间。
实时稳定性研究:将三批储存于-20±5℃条件下的申报产品经过长途高温运输后寄回,分别在申报产品生产后的第0、4、8、12、14个月检测企业参考品,结果显示:在上述各时间点检测企业参考品的各项性能指标均符合要求。因此确定申报产品的效期稳定性为:-20±5℃避光保存,有效期12个月。
三、临床评价概述
申请人在首都医科大学附属北京胸科医院、河南省人民医院和武汉市肺科医院共3家临床试验机构完成了临床试验。采用试验用体外诊断试剂与Sanger测序法及病理诊断结果进行比较研究的方法,对产品临床性能进行评价。入组病例包括肺结核、肺外结核以及肺癌、非结核分枝杆菌感染、真菌感染等其他易混淆疾病患者,样本类型为石蜡包埋组织样本。临床试验共入组受试者604例,其中阳性样本400例,阴性样本204例。试验结果显示,申报产品与Sanger测序检测阳性符合率为99.25%(95%CI:97.82%,99.85%),阴性符合率为95.59%(95%CI:91.79%,97.96%);与病理诊断对比,灵敏度为92.78%(95%CI:89.07%,95.53%),特异度为96.39%(95%CI:89.80%,99.25%)。综上所述,该产品临床试验设计符合《体外诊断试剂临床试验技术指导原则》的相关要求,临床试验结果显示该产品与对比方法一致性较好,临床性能满足临床需求。
四、产品受益风险判定
根据申请人提供的申报资料,经综合评价,在目前认知水平上,认为该产品能够较大程度地满足医疗需求,预期为适用人群带来的受益大于风险。但为保证用械安全,基于对主要剩余风险的规避,需在说明书中提示以下信息:
该产品检测结果应结合患者临床症状、体征、流行病学背景及其他临床诊断结果进行综合判断,不得作为疾病诊断的唯一标准。
综合评价意见
本申报项目为境内第三类医疗器械产品注册,属于境内同品种首个产品。申请人的注册申报资料符合现行要求,依据《医疗器械监督管理条例》(国务院令第680号)、《体外诊断试剂注册管理办法(国家食品药品监督管理总局令2014年第5号)等相关医疗器械法规与配套规章,经系统评价后,建议准予注册。