


医械创新资讯


12月1日,国家药监局发布《抗肿瘤药物的非原研伴随诊断试剂临床试验注册审查指导原则》《使用体外诊断试剂境外临床试验数据的注册审查指导原则》以及《神经和心血管手术器械-刀、剪及针注册审查指导原则》。
01/抗肿瘤药物的非原研伴随诊断试剂临床试验注册审查指导原则

伴随诊断试剂临床试验目的主要包含两个方面,一方面为确认试剂临床性能,另一方面为确认伴随诊断用途。根据伴随诊断试剂设计开发的特点,确认其伴随诊断用途的临床研究可分为如下几种情况:
(一)如为原研伴随诊断试剂,可提交该产品作为伴随诊断试剂参与的药物临床试验资料作为确认伴随诊断用途的临床试验资料,或提交与药物临床试验中所使用的CTA进行桥接试验的临床试验资料。具体可参考与抗肿瘤药物同步研发的原研伴随诊断试剂临床试验的相关要求。
(二)申报产品如为非原研伴随诊断试剂,其伴随诊断用途可根据具体情形采用下列适用的方式之一进行研究:与原研伴随诊断试剂进行一致性比对、桥接试验、已上市抗肿瘤药物疗效的观察性研究。
针对所伴随的抗肿瘤药物已上市多年、临床应用广泛、意义明确、判读易于标准化的伴随诊断试剂,如申报产品的性能与原研伴随诊断试剂具有较好的可比性,则申报产品伴随诊断用途的确认可采取与原研伴随诊断试剂进行一致性比对的方式,此类生物标志物清单见表1。申请人拟开发的未包含在表1中的生物标志物,如有需要可与监管部门充分沟通后确定其伴随诊断临床意义。
申报产品所检测的生物标志物中存在针对抗肿瘤药物疗效负性选择的生物标志物。例如RAS基因,已批准的西妥昔单抗说明书中明确载明,该药物不用于RAS基因的突变的结直肠癌患者。针对此类生物标志物,申报产品伴随诊断用途的确认可采取与原研伴随诊断试剂或CTA进行一致性比对的方式,临床试验应重点关注二者的一致性。
表1.相关生物标志物清单

注:基于当前认知及我国相关产品的开发及临床应用情况,相关抗肿瘤药物已上市、临床应用广泛、意义明确、判读易于标准化并且临床应用多年的伴随诊断生物标志物清单见表1。该清单会随着科学认知的深入及相关产品的临床应用情况适时更新。
02/使用体外诊断试剂境外临床试验数据的注册审查指导原则
本指导原则旨在为申请人使用体外诊断试剂境外临床试验数据在我国进行注册申报提供指导,适用于进行首次注册申报和相关变更注册申请的产品。
本指导原则声称的境外临床试验数据是指,全部或同期在境外具备临床试验开展所在国家(地区)要求条件的临床试验机构中,对拟在我国注册申报的体外诊断试剂进行临床试验时所产生的研究数据。
差异分析导图
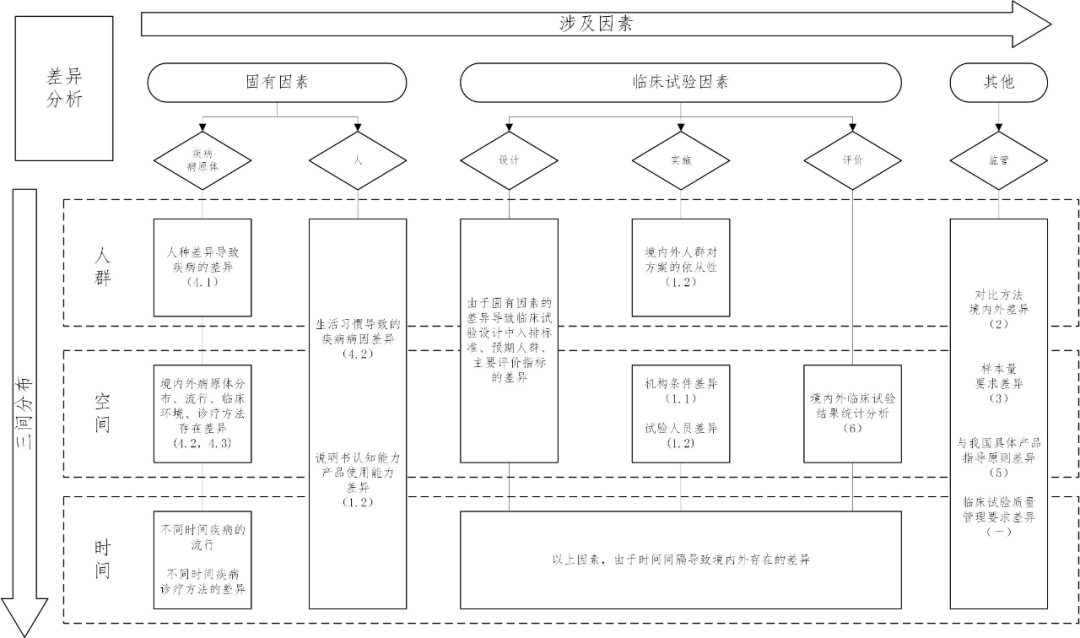
注:1.图中每部分内容均不是对该部分内容的穷举,应当根据产品的特性对境内外的差异进行具体分析。
2.不同因素、三间分布之间可能存在交叉。
3.表格主要以“(二)境内外临床试验设计关键要素的差异分析”章节为基础。
03/神经和心血管手术器械-刀、剪及针注册审查指导原则
本指导原则适用于第二类神经和心血管无源手术器械中的手术刀、手术剪及手术针。
产品技术要求
产品技术要求的制定应符合《医疗器械产品技术要求编写指南》的要求。注册申请人需根据产品的技术特征和临床使用情况来确定产品的性能指标和检验方法。对注册申请人宣称的产品的所有技术参数和功能,若适宜,均需在产品技术要求中予以规定。产品技术要求中的试验方法均需为已经过验证的方法。若对标准中的试验方法有所修改,需明确修改的内容和原因,并在研究资料中提供验证资料。对于相关行业标注、国家标准中不适用的要求条款,需说明不适用的原因和依据。
(1)产品型号/规格及其划分的说明
列表说明产品的型号、规格,明确产品的型号、规格的划分说明,鉴于该类器械型号、规格较多,建议在附录中以列表的形式提供,列表中需明确具体组件的种类和数量。
(2)性能指标
产品性能研究项目中,对于可进行客观判定的成品的功能性、安全性指标,需将其列入产品技术要求。可包括但不限于以下性能:外观、尺寸、硬度、表面粗糙度、耐腐蚀性能、连接牢固度(适用于有连接部件的产品)、使用性能(可客观判断的性能,如刀片刃口锋利度、手术剪的剪切性能等)、环氧乙烷残留量(适用于环氧乙烷灭菌的产品)、无菌(适用于灭菌状态交付的产品)。
(3)检验方法
产品的检验方法需根据性能指标设定,检验方法需优先采用公认的或已颁布的标准检验方法;自建检验方法需提供相应的方法学依据和理论基础,同时保证检验方法具有可操作性和可重现性,必要时可附相应图示进行说明,文本较大的可以附录形式提供。
(4)附录
建议注册申请人以资料性附录形式提供产品的结构图示及制造材料信息。

