《医疗器械注册与备案管理办法》(国家市场监督管理总局令第47号)和《体外诊断试剂注册与备案管理办法》(国家市场监督管理总局令第48号)(以下统称《办法》)已发布,自2021年10月1日起施行。为做好《办法》实施工作,现将有关事项通告如下:
一、关于《办法》实施前已受理注册申请项目的处理
《办法》实施前已受理但尚未作出审批决定的注册申请项目,药品监督管理部门按照原规定继续审评审批,符合上市条件的,发给医疗器械注册证。延续注册的注册证有效期起始日执行《办法》第八十四条规定。
二、关于补正材料涉及的检验报告
《办法》实施前已受理但尚未作出审批决定的注册申请项目,如补正材料涉及检验报告,注册申请人应当委托具有资质的医疗器械检验机构出具补充检验报告;如注册申请人的体系核查涵盖了检验能力,也可以按照《办法》及相关要求提交补充自检报告。
三、关于新的强制性标准实施之日前受理注册申请项目的审查
对于申请注册的医疗器械,其产品技术要求中引用的强制性标准发生变化的,除国家药监局在发布实施标准文件中另有规定外,在新标准实施之日前受理注册的,可以按照原标准进行审评审批。自新标准实施之日起,企业应当全面实施新标准,产品应当符合新标准要求。
四、关于医疗器械生物学试验
医疗器械生物学评价中涉及生物学试验的,其生物学试验报告由申请人在申请注册时作为研究资料提交。开展生物学试验,应当委托具有生物学试验资质的医疗器械检验机构按照相关标准进行试验。国外实验室出具的生物学试验报告,应当附有国外实验室表明其符合GLP实验室要求的质量保证文件。
五、关于进口医疗器械和境内生产的医疗器械注册(备案)形式
进口医疗器械,应当由境外注册申请人(备案人)申请注册(办理备案);境外企业在境内生产的医疗器械,应当由境内生产的企业作为注册申请人(备案人)申请注册(办理备案)。
六、关于第一类医疗器械备案
第一类医疗器械备案不需提交临床评价资料。
七、关于医疗器械注册管理相关文件
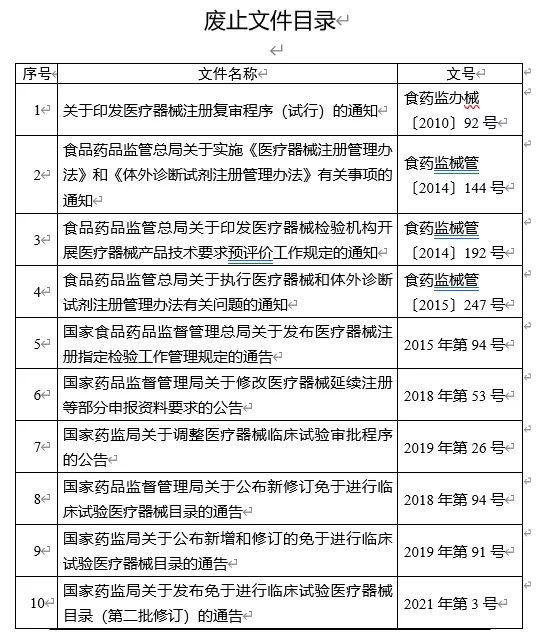
(一)《办法》实施后,附件中所列的医疗器械注册管理相关文件同时废止。
(二)《办法》中未涉及的事项,如国务院药品监督管理部门以前发布的医疗器械注册管理的文件中有明确规定的,仍执行原规定。
特此通告。
附件:废止文件目录
国家药监局
2021年9月28日