新冠抗原检测试剂最近在美国很火。
很火的原因是,根据华尔街日报9月17日的报道,美国政府开始意识到快速检测在美国复工和复学中的重要性,并意识到美国在快速新冠检测的使用上明显落后于其他国家。
很显然拜登政府也开始采取行动来弥补这一不足,在上周宣布投资20亿美金来提升快速抗原检测试剂的生产能力,并加快向社区发放免费的抗原检测试剂盒,连锁的药店也开始打折销售。
那么究竟是什么导致了美国在快速新冠抗原检测试剂上明显落后于欧洲和其他国家呢?
目前在美国只有6家IVD公司的抗原自检试剂获得FDA的EUA授权,而欧洲获得CE认证的至少有三十家,为什么?问题主要出在抗原自检试剂在美国做临床比较难。
为什么新冠抗原自检试剂在美国做临床这么难?原因主要有3个方面:
1、缺少专注医疗器械的CRO公司
2、新冠自检试剂的临床流程复杂
3、便宜且合规的临床点难找
很多的中国IVD企业家一直觉得是FDA不会批准抗原自检试剂的EUA给中国的IVD企业,其实不然,不只是中国的IVD企业在美国做临床难,美国的IVD企业做抗原自检试剂的临床也很难。FDA批准的218家分子PCR检测试剂企业中,大部分都是美国公司,可是,获得抗原自检试剂的美国企业也只有5家,主要瓶颈一样也是临床难做。
针对这3个方面的挑战,中国的IVD企业如何破解,本文将在最后介绍美国做新冠抗原自检试剂的临床流程,和临床的主要参与者(Roles):赞助方(Sponsor),调查员(Investigator),临床点管理(site manager), 临床调研员(CRA)他们的职能。
了解了抗原自检试剂的临床流程,在美国选择合适的CRO公司和临床点时,就可以少走弯路!
临床难点一:美国市场缺少专注医疗器械的CRO公司
CRO(Contract Research Organization, CRO):是研究机构外包的意思,也有人翻译成研究合同外包。主要有两类:临床CRO和药物研发CRO。
临床CRO:以接受委托临床试验(Clinical Trial)为主。主要以临床研究为主、兼顾工艺研究、注册申报、药物生产以及药物上市后再评价,也包括医疗器械的临床试验。
药物研发CRO:主要以药物发现、临床前研究为主。涉及到:PK,PD研究,一致性评价研究,活性化合物,靶向标记研究等。
在美国有超过4,000家的CRO公司,但是绝大多数CRO公司主要为药物研发和药物临床服务的,药物研究和临床的周期长,费用高。一款新药的临床一般要做10-12年,CRO公司会收取甲方非常高昂的费用。
当然,申请FDA新药批准的专业性要求也高,需要非常专业的人,最低要求是博士学历,所以CRO的人员成本相应的也很高。
而医疗器械的临床周期比较短,一般就是1-6个月,(除非是非常复杂和创新的产品),正常情况下临床难度要比开发新药低很多,从而CRO很难向甲方收很高的费用,对于CRO公司来说利润不高,
所以专注于医疗器械的临床的CRO公司数量就少,一般是大型的CRO公司顺带着做的。
大型CRO公司因为人员成本高,加上管理成本(overhead)多,基本上只有大型的药厂可以承担,或者是刚刚融了一大笔钱的Biotech公司可以承受。
如果用这样大型CRO公司做抗原自检试剂的临床,收费一定会很高。如果他们以前还没有做过类似的临床产品的经验,那收费就更高了,对他们来说,一般会组织一个团队来做,本来根本不需要一个大博士来做的工作,他们会找几个博士来做,增加了人员成本,也就不得不提高费用。
更头疼的问题是,他们的临床成功率还不一定高,用传统研究新药的思维方式,来解决一个简单OTC的消费品(Consumer Product)临床项目,就像高射炮打蚊子,效率低。
如果找一个中小型以药物开发为主的CRO公司,除非他们以前有做过和抗原自检试剂类似的OTC产品,否则,会面临同样的问题,
临床成功率也不会高,只是费用比大型CRO公司要低一点而已。
解决第一个难题的办法,是要根据FDA关于新冠抗原自检试剂的临床要求,在美国找到专业做医疗器械的CRO公司,有做过类似新冠抗原自检试剂产品的临床经验,最好是做过新冠抗原自检试剂产品的临床的CRO。
临床难点二:新冠抗原自检试剂的临床流程复杂
在2020年7月29日FDA官网公布了针对新冠抗原自检试剂的EUA指南“Template for Manufacturers ofMolecular and Antigen Diagnostic COVID-19 Tests for Non-Laboratory Use。”,其中Section. J的第8部分包含了对临床试验的要求(Clinical Evaluation),包括了:
1.临床点的要求
2.病人参与的要求
3.对比试剂的要求和临床样品的要求
4.差异性分析和解决方案
5.临床参与人数
6.临床结果计算
新冠抗原自检试剂的临床样本,和新冠分子PCR试剂以及抗原检测试剂的实验版(专业版)不一样,必须是志愿者到临床点做实地的取样和做实地的检测。专业版的样本可以使用储存的病毒样本。
在疫情期间,这个要求给临床流程增加了很大的复杂性。临床志愿者和CRO工作人员的健康安全,一直是FDA针对临床Protocol设计中最重视的一个环节,在没有疫情的情况下,对于检测试剂的临床来说这还相对容易做到,但是在新冠肆虐的当下,满足这个要求挑战就大了很多。
美国是全球新冠疫情的重灾区,截至8月28日美国已经有4317万人感染了新冠,已经有超过69万人死于新冠。
临床流程中要考虑针对志愿者在往来临床点的途中,以及在临床点的感染风险,必须要有相对应的防护措施。
另外一个挑战是志愿者的临床同意书以及她/他的新冠相关信息,都需要花更多的时间和志愿者沟通。志愿者在临床前是否有感染过新冠,是否打过疫苗,都涉及到个人隐私,如果不愿意透露,对于临床数据的采集都会有影响。
临床难点三:便宜且合规的临床点难找
在第一个难点中,我们提到大部分的美国CRO公司都是服务制药公司,那么他们自己拥有的临床点,或者他们有外包的临床点,都是围绕着长期服药和监控为主的临床过程,周期比较长,要10-12年。临床点一般都在医院的研究中心,或者是大学的研究中心,成本比较高。
医疗器械需要的临床点要求就完全不一样,需要的是短平快,在尽短的时间里招募到足够多的人,一般要在1-6个月,对于EUA的申请,更是需要在1-2个月的时间里就完成。每个志愿者的临床过程也很短,对于新冠抗原自检试剂的试验,只需要10-30分钟就结束了。
所以对于新冠抗原自检试剂临床来说,CRO公司现有的临床点,一般都不适合而且价格也太高。
CRO如果之前没有做过新冠抗原自检试剂的临床,都需要重新找临床点,而疫情给CRO公司找到合适的临床点增加了难度,FDA在2020年3月份也发布了在疫情期间选择临床点的指导文件 – “Covid19疫情期间医疗产品临床试验的指导手册”。
根据这个指导手册,临床点除了需提供满足GCP(Good Clinical Practice)的证据,还需要提供额外的志愿者安全措施,保证志愿者不能在临床的过程中感染新冠,这很显然将增加临床点的成本。
对于CRO公司来说,为了尽快取得有效的阳性案例,临床点还必须设在疫情严重的地区,这附近往往没有CRO公司,又增加了临床运营的风险和成本,很多CRO公司在疫情期间把临床点放在佛罗里达,那是一个从州长到一般民众都不带口罩的州,在这样的地方工作,东北部的CRO公司人员是不愿意的。
对于新冠抗原自检试剂的临床来说,CRO公司如何在附近找到好的临床点是减少成本和加快临床的关键。
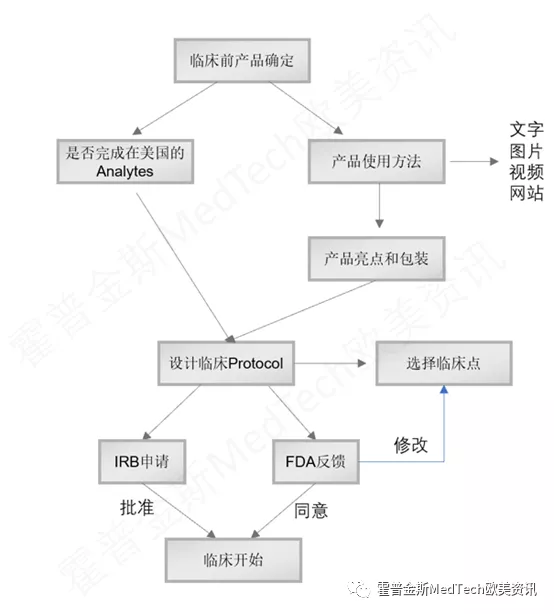
抗原自检试剂在美国的临床流程
在美国做抗原自检试剂的临床,主要分成3个步骤:
临床前的准备,申请IRB批准和FDA同意
临床的执行
完成临床报告
临床前的准备包括了以下几个关键步骤:
申请IRB(伦理委员会)批准和FDA的同意反馈是这个步骤需要完成的,也是整个临床试验最重要的两个环节。
要得到FDA的同意,这个过程中的几个重点文件是关键,包括抗原自检试剂针对用户的使用说明,针对医疗机构的说明书,这两个文件决定了FDA怎么看待这款产品,它是否符合FDA的新冠检测大方向,产品是否有创新,用户体验是否符合美国民众,这两个文件的重要性,往往被很多中国IVD企业忽视了。
临床Protocol是一个标准性的文件,一般在50-100页,需要尽量的详细,必须充分考虑FDA关于抗原自检试剂临床的要求,以及符合FDA在疫情期间对于医疗产品临床的指导要求,关键点是疫情期间志愿者的安全性如何得到保障。
在这个步骤中,FDA还需要针对临床点给出意见。
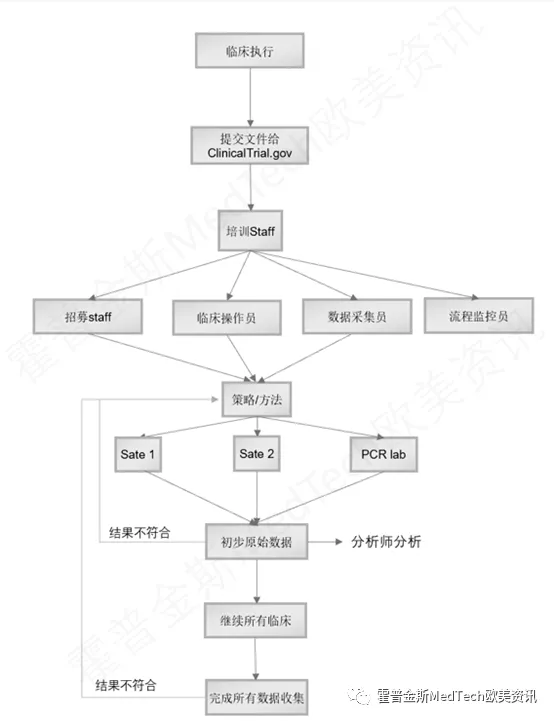
获得了FDA同意的回复和IRB的批准,临床就可以开始执行了:
在执行步骤中,关键点是对所有参与临床的CRA进行培训,然后把收集临床数据分成两个步骤,第一步先收集少量的数据,进行分析,如果数据有问题,需要立刻调整招募策略,以及检测的流程。
在完成了所有数据收集后,就需要尽快完成数据的分析,如果数据不符合FDA的要求,主要是PPA(阳性符合率)和NPA(阴性差异性)。
如果这两个数据符合要求,基本上就大功告成了,最后只需要完成第三个步骤:临床的最终报告。
虽然我们的临床功能数据结果符合了FDA的要求,FDA还是需要看到实验数据的可靠性,数据收集的流程,如果FDA需要,必须在48小时提供原始数据。
最后一步是,FDA在2021年9月23日针对新冠检测试剂EUA的新增要求,制造商必须根据最新变异病毒进行持续的临床试验,FDA有权要求制造商在48小时内提供最新变异病毒的临床数据,当然如果没有新的变异病毒,这个要求是不适用的。
关于不同临床人员的角色和职能,在这里我们需要强调一下Investigator的重要性。所有的临床人员里,Investigator是最关键的一个角色,也是FDA在审核临床Protocol和最后的临床报告时,最关键的一个人,是的,Investigator必须是个人,不能是机构。Investigator的教育背景,工作经验,合规培训背景等,都是FDA在审批临床Protocol时关注的。
Investigator的主要作用是统筹整个临床试验的执行,必须严格按照临床Protocol的流程来操作,任何调整都必须获得Sponsor的同意,并且需要修改Protocol和获得IRB的同意。
保证临床志愿者的安全是Investigator最重要的职责之一,同时要保证临床试验点的运营符合GCP的规范,最新FDA法规和指南。
在执行临床之前,Investigator负责所有临床人员的招募和培训,志愿者的招募和面试,监督临床试验的流程符合Protocol等。
对于希望在美国申请新冠抗原检测试剂临床试验的IVD企业,以上的临床流程介绍对你们在美国选择合适的CRO公司完成临床能有所帮助。